














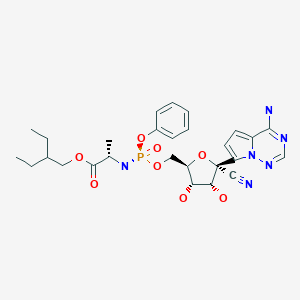
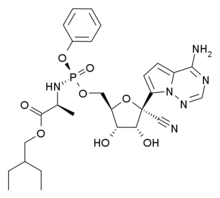
Remdesivir
| Formula |
C27H35N6O8P
|
|---|---|
| CAS |
1809249-37-3
|
| Mol weight |
602.576
|
レムデシビル
L-Alanine, N-((S)-hydroxyphenoxyphosphinyl)-, 2-ethylbutyl ester, 6-ester with 2-C-(4-aminopyrrolo(2,1-f)(1,2,4)triazin-7-yl)-2,5-anhydro-D-altrononitrile
2-Ethylbutyl (2S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo(2,1-f)(1,2,4)triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propanoate
- 2-Ethylbutyl (2S)-2-[[(S)-[[(2R,3S,4R,5R)-5-(4-aminopyrrolo(2,1-f)(1,2,4)triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl]methoxy]phenoxyphosphoryl]amino]propanoate
- 2-Ethylbutyl (2S)-2-[[[[(2R,3S,4R,5R)-5-(4-aminopyrrolo(2,1-f)(1,2,4)triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl]methoxy]phenoxyphosphoryl]amino]propanoate
- 2-Ethylbutyl N-[(S)-[2-C-(4-aminopyrrolo(2,1-f)(1,2,4)triazin-7-yl)-2,5-anhydro-D-altrononitril-6-O-yl]phenoxyphosphoryl]-L-alaninate
- GS 5734
- L-Alanine, N-[(S)-hydroxyphenoxyphosphinyl)-, 2-ethylbutyl ester,6-ester with 2-C-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-2,5-anhydro-D-altrononitrile
Treatment of viral infections
Phase III, clinical trials for the treatment of hospitalized patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (COVID-19). National Institute of Allergy and Infectious Diseases (NIAID) is evaluating remdesivir in phase II/III clinical trials for the treatment of Ebola virus infection.
The compound has been evaluated in preclinical studies for the potential treatment of Middle East respiratory syndrome coronavirus (MERS-CoV) and severe acute respiratory syndrome coronavirus (SARS-CoV) infections.
Remdesivir is a nucleoside analogue, with effective antiviral activity, with EC50s of 74 nM for ARS-CoV and MERS-CoV in HAE cells, and 30 nM for murine hepatitis virus in delayed brain tumor cells.
Remdesivir (development code GS-5734) is a novel antiviral drug in the class of nucleotide analogs. It was developed by Gilead Sciences as a treatment for Ebola virus disease and Marburg virus infections,[1] though it has subsequently also been found to show antiviral activity against other single stranded RNA viruses such as respiratory syncytial virus, Junin virus, Lassa fever virus, Nipah virus, Hendra virus, and the coronaviruses (including MERS and SARS viruses).[2][3] It is being studied for SARS-CoV-2 and Nipah and Hendra virus infections.[4][5][6] Based on success against other coronavirus infections, Gilead provided remdesivir to physicians who treated an American patient in Snohomish County, Washington in 2020, infected with SARS-CoV-2[7] and is providing the compound to China to conduct a pair of trials in infected individuals with and without severe symptoms.[8]
Research usage
Laboratory tests suggest remdesivir is effective against a wide range of viruses, including SARS-CoV and MERS-CoV. The medication was pushed to treat the West African Ebola virus epidemic of 2013–2016. Although the drug turned out to be safe, it was not particularly effective against filoviruses such as the Ebola virus.
Ebola virus
Remdesivir was rapidly pushed through clinical trials due to the West African Ebola virus epidemic of 2013–2016, eventually being used in at least one human patient despite its early development stage at the time. Preliminary results were promising and it was used in the emergency setting during the Kivu Ebola epidemic that started in 2018 along with further clinical trials, until August 2019, when Congolese health officials announced that it was significantly less effective than monoclonal antibody treatments such as mAb114 and REGN-EB3. The trials, however, established its safety profile.[9][10][11][12][13][14][15][16]
SARS-CoV-2
In response to the 2019–20 coronavirus outbreak induced by coronavirus SARS-CoV-2, Gilead provided remdesivir for a “small number of patients” in collaboration with Chinese medical authorities for studying its effects.[17]
Gilead also started laboratory testing of remdesivir against SARS-CoV-2. Gilead stated that remdesivir was “shown to be active” against SARS and MERS in animals.[3][18]
In late January 2020, remdesivir was administered to the first US patient to be confirmed to be infected by SARS-CoV-2, in Snohomish County, Washington, for “compassionate use” after he progressed to pneumonia. While no broad conclusions were made based on the single treatment, the patient’s condition improved dramatically the next day,[7] and he was eventually discharged.[19]
Also in late January 2020, Chinese medical researchers stated to the media that in exploratory research considering a selection of 30 drug candidates. Remdesivir and two other drugs, chloroquine and lopinavir/ritonavir, seemed to have “fairly good inhibitory effects” on SARS-CoV-2 at the cellular level. Requests to start clinical testing were submitted,[20][21]. On February 6, 2020, a clinical trial of remdesivir began in China.[22]
Other viruses
The active form of remdesivir, GS-441524, shows promise for treating feline coronavirus.[23]
Mechanism of action and resistance
Remdesivir is a prodrug that metabolizes into its active form GS-441524. GS-441524 is an adenosine nucleotide analog that confuses viral RNA polymerase and evades proofreading by viral exoribonuclease (ExoN), causing a decrease in viral RNA production. It was unknown whether it terminates RNA chains or causes mutations in them.[24]However, it has been learned that the RNA dependent RNA polymerase of ebolavirus is inhibited for the most part by delayed chain termination.[25]
Mutations in the mouse hepatitis virus RNA replicase that cause partial resistance were identified in 2018. These mutations make the viruses less effective in nature, and the researchers believe they will likely not persist where the drug is not being used.[24]
MORE SYNTHESIS COMING, WATCH THIS SPACE…………………..

SYNTHESIS
Remdesivir can be synthesized in multiple steps from ribose derivatives. The figure below is one of the synthesis route of remdesivir invented by Chun et al. from Gilead Sciences.[26]In this method, intermediate a is firstly prepared from L-alanine and phenyl phosphorodichloridate in presence of triethylamine and dichloromethane; triple benzyl-protected ribose is oxidized by dimethyl sulfoxide with acetic anhydride and give the lactone intermediate b; pyrrolo[2,1-f][1,2,4]triazin-4-amine is brominated, and the amine group is protected by excess trimethylsilyl chloride. n-Butyllithium undergoes a halogen-lithium exchange reaction with the bromide at -78 °C to yield the intermediate c. The intermediate b is then added to a solution containing intermediate c dropwise. After quenching the reaction in a weakly acidic aqueous solution, a mixture of 1: 1 anomers was obtained. It was then reacted with an excess of trimethylsilyl cyanide in dichloromethane at -78 °C for 10 minutes. Trimethylsilyl triflate was added and reacts for an additional 1 hour, and the mixture was quenched in an aqueous sodium hydrogen carbonate. A nitrile intermediate was obtained. The protective group, benzyl, was then removed with boron trichloride in dichloromethane at -20 °C. The excess of boron trichloride was quenched in a mixture of potassium carbonate and methanol. A benzyl-free intermediate was obtained. The isomers were then separated via reversed-phase HPLC. The optically pure compound and intermediate a are reacted with trimethyl phosphate and methylimidazole to obtain a diastereomer mixture of remdesivir. In the end, optically pure remdesivir can be obtained through methods such as chiral resolution.

PATENT
WO 2018204198
Prevention and treatment methods for some Arenaviridae , Coronaviridae , Filoviridae, Flaviviridae, and Paramyxoviridae viruses present challenges due to a lack of vaccine or post-exposure treatment modality for preventing or managing these infections. In some cases, patients only receive supportive and resource intensive therapy such as electrolyte and fluid balancing, oxygen, blood pressure maintenance, or treatment for secondary infections. Thus, there is a need for antiviral therapies having a potential for broad antiviral activity.
[0004] The compound (S)-2-ethylbutyl 2-(((S)-(((2R,3 S,4R,5R)-5-(4-aminopyrrolo[2, 1-f][l,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy) phosphoryl)amino)propanoate, referred herein as Compound 1 or Formula I, is known to exhibit antiviral properties against Arenaviridae, Coronaviridae, Filoviridae, and
Paramyxoviridae viruses as described in Warren, T. et al., Nature (2016) 531 :381-385 and antiviral activities against Flaviviridae viruses as described in co-pending United States provisional patent application no. 62/325,419 filed April 20, 2016.
[0005] (S)-2-Ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2, l-f][l,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)
propanoate or 2-ethylbutyl ((S)-(((2R,3 S,4R,5R)-5-(4-aminopyrrolo[2, l-f][l,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)-L-alaninate, (Formula I), has the following structure:
Formula I
PATENT
WO 2017184668
https://patents.google.com/patent/WO2017184668A1/en
A. Preparation of Compounds
Example 1. (2S)-ethyl 2-(chloro(phenoxy)phosphorylamino)pro anoate (Chloridate A)

[0246] Ethyl alanine ester hydrochloride salt (1.69 g, 11 mmol) was dissolved in anhydrous CH2CI2 (10 mL) and the mixture stirred with cooling to 0 °C under N2(g). Phenyl dichlorophosphate (1.49 mL, 10 mmol) was added followed by dropwise addition of Et3N over 10 min. The reaction mixture was then slowly warmed to RT and stirred for 12 h. Anhydrous Et20 (50 mL) was added and the mixture stirred for 30 min. The solid that formed was removed by filtration, and the filtrate concentrated under reduced pressure. The residue was subjected to silica gel chromatography eluting with 0-50% EtOAc in hexanes to provide intermediate A (1.13 g, 39%). H NMR (300 MHz, CDC13) δ 7.39-7.27 (m, 5H), 4.27 (m, 3H), 1.52 (m, 3H), 1.32 (m, 3H). 31P NMR (121.4 MHz, CDC13) δ 8.2, 7.8.
Example 2. (2S)-2-ethylbutyl 2-(chloro(phenoxy)phosphorylamino)propanoate
(Chloridate B

[0247] The 2-ethylbutyl alanine chlorophosphoramidate ester B was prepared using the same procedure as chloridate A except substituting 2-ethylbutyl alanine ester for ethyl alanine ester. The material is used crude in the next reaction. Treatment with methanol or ethanol forms the displaced product with the requisite LCMS signal.
Example 3. (2S)-isopropyl 2-(chloro(phenoxy)phosphorylamino)propanoate
(Chloridate C)

C
[0248] The isopropyl alanine chlorophosphoramidate ester C was prepared using the same procedure as chloridate A except substituting isopropyl alanine ester for the ethyl alanine ester. The material is used crude in the next reaction. Treatment with methanol or ethanol forms the displaced product with the requisite LCMS signal.
Example 4. (2S)-2-ethylbutyl 2-((((2R,3S,4R,5R)-5-(4-aminopyrrolo[l,2-firi,2,41triazin- 7-yl)-5-cvano-3,4-dihvdroxytetrahydrofuran-2- yl)methoxy)(phenoxy)phosphorylamino)propanoate (Compound 9)
[0249] Compound 9 can be prepared by several methods described below. Procedure 1

[0250] Prepared from Compound 1 and chloridate B according to the same method as for the preparation of compound 8 as described in PCT Publication no. WO 2012/012776. 1H NMR (300 MHz, CD3OD) δ 7.87 (m, 1H), 7.31-7.16 (m, 5H), 6.92-6.89 (m, 2H), 4.78 (m, 1H), 4.50-3.80 (m, 7H), 1.45-1.24 (m, 8H), 0.95-0.84 (m, 6H). 31P NMR (121.4 MHz, CD3OD) δ 3.7. LCMS m/z 603.1 [M+H], 601.0 [M-H].
Procedure 2

9
[0251] (2S)-2-ethylbutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,l-f][l,2,4]triazin-7- yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino) propanoate. (2S)-2-ethylbutyl 2-(((4-nitrophenoxy)(phenoxy)phosphoryl)amino)propanoate (1.08 g, 2.4 mmol) was dissolved in anhydrous DMF (9 mL) and stirred under a nitrogen atmosphere at RT. (2R,3R,4S,5R)-2-(4-aminopyrrolo[2,l-f][l,2,4]triazin-7-yl)-3,4- dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile (350 mg, 1.2 mmol) was added to the reaction mixture in one portion. A solution of i-butylmagnesium chloride in THF (1M, 1.8 mL, 1.8 mmol) was then added to the reaction drop wise over 10 minutes. The reaction was stirred for 2 h, at which point the reaction mixture was diluted with ethyl acetate (50 mL) and washed with saturated aqueous sodium bicarbonate solution (3 x 15 mL) followed by saturated aqueous sodium chloride solution (15 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting oil was purified with silica gel column chromatography (0-10% MeOH in DCM) to afford (2S)-2- ethylbutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2, l-f][l,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino) propanoate (311 mg, 43%, 1 :0.4 diastereomeric mixture at phosphorus) as a white solid. H NMR (400 MHz, CD3OD) δ 7.85 (m, 1H), 7.34 – 7.23 (m, 2H), 7.21 – 7.09 (m, 3H), 6.94 – 6.84 (m, 2H), 4.78 (d, / = 5.4 Hz, 1H), 4.46 – 4.33 (m, 2H), 4.33 – 4.24 (m, 1H), 4.18 (m, 1H), 4.05 – 3.80 (m, 3H), 1.52 – 1.39 (m, 1H), 1.38 – 1.20 (m, 7H), 0.85 (m, 6H). 31P NMR (162 MHz, CD3OD) δ 3.71, 3.65. LCMS m/z 603.1 [M+H], 600.9 [M-H]. HPLC (2-98% MeCN-H20 gradient with 0.1% TFA modifier over 8.5 min, 1.5mL/min, Column: Phenomenex Kinetex C18, 2.6 um 100 A, 4.6 x 100 mm ) tR = 5.544 min, 5.601 min
Separation of the (S) and (R) Diastereomers
[0252] (2S)-2-ethylbutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,l-f][l,2,4]triazin-7-yl)- 5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino) propanoate was dissolved in acetonitrile. The resulting solution was loaded onto Lux Cellulose-2 chiral column, equilibrated in acetonitrile, and eluted with isocratic
acetonitrile/methanol (95 :5 vol/vol). The first eluting diastereomer had a retention time of 17.4 min, and the second eluting diastereomer had a retention time of 25.0 min.
[0253] First Eluting Diastereomer is (S)-2-ethylbutyl 2-(((R)-(((2R,3S,4R,5R)-5-(4- aminopyrrolo[2, 1 -f] [ 1 ,2,4]triazin-7-yl)-5-cyano-3 ,4-dihydroxytetrahydrofuran-2- yl)methoxy)(phenoxy)phos horyl)amino)propanoate:

!HNMR (400 MHz, CD3OD) δ 8.05 (s, 1H), 7.36 (d, / = 4.8 Hz, 1H), 7.29 (br t, J = 7.8 Hz, 2H), 7.19 – 7.13 (m, 3H), 7.11 (d, / = 4.8 Hz, 1H), 4.73 (d, / = 5.2 Hz, 1H), 4.48 – 4.38 (m, 2H), 4.37 – 4.28 (m, 1H), 4.17 (t, / = 5.6 Hz, 1H), 4.08 – 3.94 (m, 2H), 3.94 – 3.80 (m, 1H), 1.48 (sep, / = 12.0, 6.1 Hz, 1H), 1.34 (p, / = 7.3 Hz, 4H), 1.29 (d, / = 7.2 Hz, 3H), 0.87 (t, / = 7.4 Hz, 6H). 31PNMR (162 MHz, CD3OD) δ 3.71 (s). HPLC (2-98% MeCN-H20 gradient with 0.1 % TFA modifier over 8.5 min, 1.5mL/min, Column: Phenomenex Kinetex C18, 2.6 um 100 A, 4.6 x 100 mm ) is = 5.585 min. [0254] Second Eluting Diastereomer is (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4- aminopyrrolo[2, 1 -f] [ 1 ,2,4]triazin-7-yl)-5-cyano-3 ,4-dihydroxytetrahydrofuran-2- yl)methoxy)(phenoxy)phosphoryl)amino)propanoate:

HNMR (400 MHz, CD3OD) δ 8.08 (s, 1H), 7.36 – 7.28 (m, 3H), 7.23 – 7.14 (m, 3H), 7.08 (d, 7 = 4.8 Hz, 1H), 4.71 (d, 7 = 5.3 Hz, 1H), 4.45 – 4.34 (m, 2H), 4.32 – 4.24 (m, 1H), 4.14 (t, / = 5.8 Hz, 1H), 4.08 – 3.94 (m, 2H), 3.93 – 3.85 (m, 1H), 1.47 (sep, / = 6.2 Hz, 1H), 1.38 – 1.26 (m, 7H), 0.87 (t, / = 7.5 Hz, 6H). 31PNMR (162 MHz, CD3OD) δ 3.73 (s). HPLC (2- 98% MeCN-H20 gradient with 0.1% TFA modifier over 8.5 min, 1.5mL/min, Column: Phenomenex Kinetex C18, 2.6 urn 100 A, 4.6 x 100 mm ) tR = 5.629 min.
Example 5. (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolor2J- f|[l,2,41triazin-7-yl)-5-cvano-3,4-dihvdroxytetrahvdrofuran-2- yl)methoxy)(phenoxy)phosphoryl)amino)propanoate (32)

[0255] The preparation of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,l f][l,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2- yl)methoxy)(phenoxy)phosphoryl)amino)propanoate is described below.
Preparation of (3R,4R,5R)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl)dihydrofuran-2(3H)- one.

[0256] (3R,4R,5R)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-ol (15.0g) was combined with MTBE (60.0 mL), KBr (424.5 mg), aqueous K2HP04solution (2.5M, 14.3 mL), and TEMPO (56 mg). This mixture was cooled to about 1 °C. Aqueous bleach solution (7.9%wt.) was slowly charged in portions until complete consumption of starting material as indicated through a starch/iodide test. The layers were separated, and the aqueous layer was extracted with MTBE. The combined organic phase was dried over MgS04 and concentrated under reduced pressure to yield the product as a solid.
Preparation (4-amino-7-iodopyrrolor2,l-fl ri,2,41triazine)

[0257] To a cold solution of 4-aminopyrrolo[2, l-f][l,2,4]-triazine (10.03 g; 74.8 mmol) in N,N-dimethylformamide (70.27 g), N-iodosuccinimide (17.01g; 75.6 mmol) was charged in portions, while keeping the contents at about 0 °C. Upon reaction completion (about 3 h at about 0 °C), the reaction mixture was transferred into a 1 M sodium hydroxide aqueous solution (11 g NaOH and 276 mL water) while keeping the contents at about 20-30 °C. The resulting slurry was agitated at about 22 °C for 1.5 h and then filtered. The solids are rinsed with water (50 mL) and dried at about 50 °C under vacuum to yield 4-amino-7- iodopyrrolo[2,l-f] [l,2,4]triazine as a solid. !H NMR (400 MHz, DMSO-d6) δ 7.90 (s, 1H), 7.78 (br s, 2H), 6.98 (d, J = 4.4 Hz, 1H), 6.82 (d, J = 4.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 155.7, 149.1, 118.8, 118.1, 104.4, 71.9. MS m/z = 260.97 [M+H].
Preparation (3R,4R,5R)-2-(4-aminopyrrolor2, l-firi,2,41triazin-7-yl)-3,4-bis(benzyloxy)-5- ((benzyloxy)methyl)tetrahvdrofuran-2-ol via (4-amino-7-iodopyrrolor2,l-fl ri,2,41triazine)

[0258] To a reactor under a nitrogen atmosphere was charged iodobase 2 (81 g) and THF (1.6 LV). The resulting solution was cooled to about 5 °C, and TMSC1 (68 g) was charged. PhMgCl (345mL, 1.8 M in THF) was then charged slowly while maintaining an internal temperature at about < 5°C. The reaction mixture was stirred at about 0°C for 30 min, and then cooled to about -15 °C. z‘PrMgCl-LiCl (311 mL, 1.1 M in THF) was charged slowly while maintaining an internal temperature below about -12 °C. After about 10 minutes of stirring at about -15 °C, the reaction mixture was cooled to about -20 °C, and a solution of lactone 1 (130 g) in THF (400 mL) was charged. The reaction mixture was then agitated at about -20 °C for about 1 h and quenched with AcOH (57 mL). The reaction mixture was warmed to about 0 °C and adjusted to pH 7-8 with aqueous NaHCC>3 (5 wt%, 1300 mL). The reaction mixture was then diluted with EtOAc (1300 mL), and the organic and aqueous layers were separated. The organic layer was washed with IN HC1 (1300 mL), aqueous NaHCC>3 (5 wt%, 1300 mL), and brine (1300 mL), and then dried over anhydrous Na2S04 and concentrated to dryness. Purification by silica gel column chromatography using a gradient consisting of a mixture of MeOH and EtOAc afforded the product.
Preparation ((2S)-2-ethylbutyl 2- (((perfluorophenoxy)(phenoxy)phosphoryl)amino)propanoate) (mixture of Sp and Rp):
1 ) phenyl dichlorophosphate
CH2CI2, -78 °C to ambient
2) pentafluorophenol
Et3N, 0 °C to ambient

[0259] L- Alanine 2-ethylbutyl ester hydrochloride (5.0 g, 23.84 mmol) was combined with methylene chloride (40 mL), cooled to about -78 °C, and phenyl dichlorophosphate (3.65 mL, 23.84 mmol) was added. Triethylamine (6.6 mL, 47.68 mmol) was added over about 60 min at about -78 °C and the resulting mixture was stirred at ambient temperature for 3h. The reaction mixture was cooled to about 0 °C and pentafluorophenol (4.4 g, 23.84 mmol) was added. Triethylamine (3.3 mL, 23.84 mmol) was added over about 60 min. The mixture was stirred for about 3h at ambient temperature and concentrated under reduced pressure. The residue was dissolved in EtOAc, washed with an aqueous sodium carbonate solution several times, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using a gradient of EtOAc and hexanes (0 to 30%). Product containing fractions were concentrated under reduced pressure to give (2S)-2-ethylbutyl 2-(((perfluorophenoxy)(phenoxy)phosphoryl)amino)propanoate as a solid. H NMR (400 MHz, Chloroform-d) δ 7.41 – 7.32 (m, 4H), 7.30 – 7.17 (m, 6H), 4.24 – 4.16 (m, 1H), 4.13 – 4.03 (m, 4H), 4.01 – 3.89 (m, 1H), 1.59 – 1.42 (m, 8H), 1.40 – 1.31 (m, 8H), 0.88 (t, J = 7.5 Hz, 12H). 31P NMR (162 MHz, Chloroform-d) δ – 1.52. 19F NMR (377 MHz, Chloroform-d) δ – 153.63, – 153.93 (m), – 160.05 (td, J = 21.9, 3.6 Hz), – 162.65 (qd, J = 22.4, 20.5, 4.5 Hz). MS m/z = 496 [M+H]. Preparation of Title Compound (mixture of Sp and Rp):
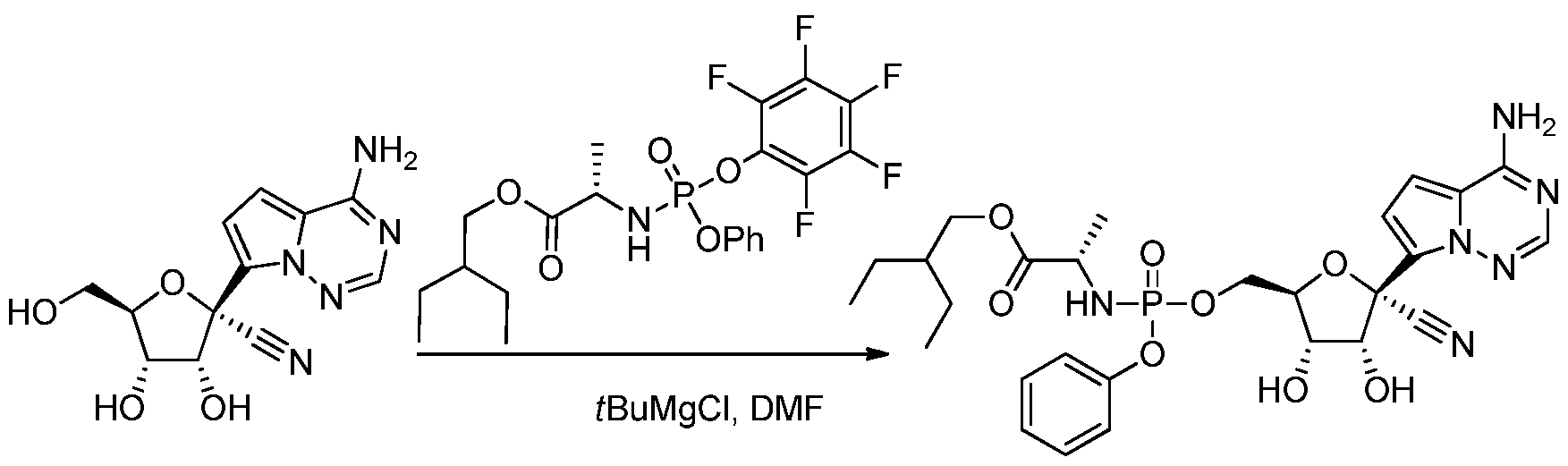
[0260] The nucleoside (29 mg, 0.1 mmol) and the phosphonamide (60 mg, 0.12 mmol) and N,N-dimethylformamide (2 mL) were combined at ambient temperature. 7¾ri-Butyl magnesiumchloride (1M in THF, 0.15 mL) was slowly added. After about lh, the reaction was diluted with ethyl acetate, washed with aqueous citric acid solution (5%wt.), aqueous saturated NaHC03 solution and saturated brine solution. The organic phase was dried over Na2S04 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using a gradient of methanol and CH2CI2 (0 to 5%). Product containing fractions were concentrated under reduced pressure to provide the product.
Preparation of (3aR,4R,6R,6aR)-4-(4-aminopyrrolor2, l-firi,2,41triazin-7-yl)-6- (hvdroxymethyl)-2,2-dimethyltetrahydrofuror3,4-diri,31dioxole-4-carbonitrile:

[0261] To a mixture of (2R,3R,4S,5R)-2-(4-aminopyrrolo[2, l-f] [l,2,4]triazin-7-yl)-3,4- dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile (5.8g, 0.02 mol), 2,2- dimethoxypropane (11.59 mL, 0.09 mol) and acetone (145 mL) at ambient temperature was added sulfuric acid (18M, 1.44 mL). The mixture was warmed to about 45 °C. After about 30 min, the mixture was cooled to ambient temperature and sodium bicarbonate (5.8 g) and water 5.8 mL) were added. After 15 min, the mixture was concentrated under reduced pressure. The residue was taken up in ethyl acetate (150 mL) and water (50 mL). The aqueous layer was extracted with ethyl acetate (2 x 50 mL). The combined organic phase was dried over sodium sulfate and concentrated under reduced pressure to give crude (2R,3R,4S,5R)-2-(4-aminopyrrolo[2, l-f] [l,2,4]triazin-7-yl)-3,4-dihydroxy-5- (hydroxymethyl)tetrahydrofuran-2-carbonitrile. !H NMR (400 MHz, CD3OD) δ 7.84 (s, 1H), 6.93 (d, / = 4.6 Hz, 1H), 6.89 (d, / = 4.6 Hz, 1H), 5.40 (d, / = 6.7 Hz, 1H), 5.00 (dd, / = 6.7, 3.3 Hz, 1H), 4.48 – 4.40 (m, 1H), 3.81 – 3.72 (m, 2H), 1.71 (s, 3H), 1.40 (s, 3H). MS m/z = 332.23 [M+l].
Preparation of (2S)-2-ethylbutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolor2,l-firi,2,41triazin- 7-yl)-5-cvano-3,4-dihvdroxytetrahydrofuran-2- yl)methoxy)(phenoxy)phosphoryl)amino)propanoate:

[0262] Acetonitrile (100 mL) was combined with (2S)-2-ethylbutyl 2-(((4- nitrophenoxy)(phenoxy)phosphoryl)-amino)propanoate (9.6 g, 21.31 mmol), the substrate alcohol (6.6 g, 0.02 mol), magnesium chloride (1.9 g, 19.91 mmol) at ambient temperature. The mixture was agitated for about 15 min and N,N-diisopropylethylamine (8.67 mL, 49.78 mmol) was added. After about 4h, the reaction was diluted with ethyl acetate (100 mL), cooled to about 0 °C and combined with aqueous citric acid solution (5%wt., 100 mL). The organic phase was washed with aqueous citric acid solution (5%wt., 100 mL) and aqueous saturated ammonium chloride solution (40 mL), aqueous potassium carbonate solution
(10%wt., 2 x 100 mL), and aqueous saturated brine solution (100 mL). The organic phase was dried with sodium sulfate and concentrated under reduced pressure to provide crude product. !H NMR (400 MHz, CD3OD) δ 7.86 (s, 1H), 7.31 – 7.22 (m, 2H), 7.17 – 7.09 (m, 3H), 6.93 – 6.84 (m, 2H), 5.34 (d, / = 6.7 Hz, 1H), 4.98 (dd, / = 6.6, 3.5 Hz, 1H), 4.59 – 4.50 (m, 1H), 4.36 – 4.22 (m, 2H), 4.02 (dd, / = 10.9, 5.7 Hz, 1H), 3.91 (dd, / = 10.9, 5.7 Hz, 1H), 3.83 (dq, / = 9.7, 7.1 Hz, 1H), 1.70 (s, 3H), 1.50 – 1.41 (m, 1H), 1.39 (s, 3H), 1.36 – 1.21 (m, 7H), 0.86 (t, / = 7.4 Hz, 6H). MS m/z = 643.21 [M+l]. Preparation of (S)-2-ethylbutyl 2-(((S)-(((2R.3S.4R.5R)-5-(4-aminopyrrolor2.1- firi,2,41triazin-7-yl)-5-cvano-3,4-ditivdroxytetratiydrofuran-2- yl)methoxy)( henoxy)phosphoryl)amino)propanoate (Compound 32)

Compound 32
[0263] The crude acetonide (12.85 g) was combined with tetrahydrofuran (50 mL) and concentrated under reduced pressure. The residue was taken up in tetrahydrofuran (100 mL), cooled to about 0 °C and concentrated HC1 (20 mL) was slowly added. The mixture was allowed to warm to ambient temperature. After consumption of the starting acetonide as indicated by HPLC analysis, water (100 mL) was added followed by aqueous saturated sodium bicarbonate solution (200 mL). The mixture was extracted with ethyl acetate (100 mL), the organic phase washed with aqueous saturated brine solution (50 mL), dried over sodium sulfated and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using a gradient of methanol and ethyl acetate (0 to 20%).
Product containing fractions were concentrated under reduced pressure to provide the product.
PATENT
US 20170071964
US 20160122374
PAPER
Journal of Medicinal Chemistry (2017), 60(5), 1648-1661.
https://pubs.acs.org/doi/full/10.1021/acs.jmedchem.6b01594

The recent Ebola virus (EBOV) outbreak in West Africa was the largest recorded in history with over 28,000 cases, resulting in >11,000 deaths including >500 healthcare workers. A focused screening and lead optimization effort identified 4b (GS-5734) with anti-EBOV EC50 = 86 nM in macrophages as the clinical candidate. Structure activity relationships established that the 1′-CN group and C-linked nucleobase were critical for optimal anti-EBOV potency and selectivity against host polymerases. A robust diastereoselective synthesis provided sufficient quantities of 4b to enable preclinical efficacy in a non-human-primate EBOV challenge model. Once-daily 10 mg/kg iv treatment on days 3–14 postinfection had a significant effect on viremia and mortality, resulting in 100% survival of infected treated animals [ Nature 2016, 531, 381−385]. A phase 2 study (PREVAIL IV) is currently enrolling and will evaluate the effect of 4b on viral shedding from sanctuary sites in EBOV survivors.
(S)-2-Ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propanoate (4b)
PAPER
Nature (London, United Kingdom) (2016), 531(7594), 381-385.
https://www.nature.com/articles/nature17180
 |
|
| Clinical data | |
|---|---|
| Other names | GS-5734 |
| Routes of administration |
By mouth, insufflation |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C27H35N6O8P |
| Molar mass | 602.585 g·mol−1 |
| 3D model (JSmol) | |
|
|
| Clinical data | |
|---|---|
| Other names | GS-5734 |
| Routes of administration |
By mouth, insufflation |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C27H35N6O8P |
| Molar mass | 602.585 g·mol−1 |
| 3D model (JSmol) | |
//////////////Remdesivir, レムデシビル , UNII:3QKI37EEHE, ремдесивир , ريمديسيفير , 瑞德西韦 , GS-5734 , GS 5734, PHASE 3 , CORONOVIRUS, COVID-19
CCC(CC)