














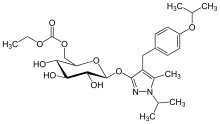
REMOGLIFLOZIN ETABONATE
5-methyl-4-[4-(1-methylethoxy)benzyl]-1-(1-methylethyl)-1H-pyrazol-3-yl 6-O-(ethoxycarbonyl)-β-D-glucopyranoside,
CAS 442201-24-3
189075 BHV-091009 GSK-189075 GSK-189075A KGT-1681
BHV Pharma Kissei (Originator) , GlaxoSmithKline
Remogliflozin etabonate
Phase II
A SGLT-2 antagonist potentially for the treatment of type 2 diabetes, non-alcoholic steatohepatitis (NASH), obesity.
![]()
GSK-189075; GSK-189075A
- Molecular FormulaC26H38N2O9
- Average mass522.588
Remogliflozin etabonate (INN/USAN)[1] is a proposed drug of the gliflozin class for the treatment of non-alcoholic steatohepatitis (“NASH”) and type 2 diabetes. Remogliflozin is being developed by Avolynt, Inc.[2]
Remogliflozin etabonate, also known as GSK 189075A or GSK 189075, is a SGLT2 inhibitor under development for the treatment of type 2 diabetes. Remogliflozin etabonate is a pro-drug of remogliflozin. Remogliflozin inhibits the sodium-glucose transport proteins (SGLT), which are responsible for glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine. Remogliflozin is selective for SGLT2.
Remogliflozin etabonate also known as 5-methyl-4-[4-(1-methylethoxy)benzyl]-1-(1- methylethyl)-1H-pyrazol-3-yl 6-0-(ethoxycar onyl)-β-D-glucopyranoside of the following formula
(«):

(I)
Another nomenclature convention provides this molecule as 3-(6-0-ethoxycarbonyl-.beta.-D- glucopyranosyloxy)-4-[(4-isopropoxyphenyl)methyl]-1-isopropyl-5-methylpyrazole. Remogliflozin etabonate is also known as GSK 189075 or KGT-1681. Salts of compounds of formula (i) are useful as the active ingredient in the pharmaceutical presentation of the invention. Such salts may be as described in US Patent 7,084,123 issued August 1, 2006, herein incorporated by reference. Examples of such salts include acid addition salts with mineral acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid and the like, acid addition salts with organic acids such as formic acid, acetic acid,
methanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, propionic acid, citric acid, succinic acid, tartaric acid, fumaric acid, butyric acid, oxalic acid, malonic acid, maieic acid, lactic acid, malic acid, carbonic acid, glutamic acid, aspartic acid, adipic acid, oleic acid, stearic acid and the like, and salts with inorganic bases such as a sodium salt, a potassium salt, a calcium salt, a magnesium salt and the like.
The compounds represented by the above formula (I) include their solvates with pharmaceutically acceptable solvents such as ethanol and water.
Remogliflozin etabonate may be prepared as described in US Patents 7,084,123 and 7,375,087, in particular Example 1 of US Patent 7,084,123, each herein incorporated by reference.
Remogliflozin etabonate is the pro-drug of remogliflozin (also known as GSK189074 or KGT-1650).
Remogliflozin etabonate has the potential to be used as monotherapy for the treatment of T2DM. To date, studies have assessed the efficacy, safety and tolerability up to 12 weeks, with varying efficacy so there is a need to characterize the profile of a number of selected formulated doses over a 12-week period. The study is designed with a placebo treatment arm to enable the profile of the drug to be further characterized and for maximal glycemic effect to be achieved. However to minimize the time which subjects may have sub-optimal glycemic control, the double blind study medication has been limited to 12 weeks duration. In addition, criteria have been included to allow the introduction of rescue therapy after 6 weeks for those subjects who have a high FP.
Diabetes mellitus is a serious disease afflicting over 100 million people worldwide. In the United States, there are more than 12 million diabetics, with 600,000 new cases diagnosed each year.
Diabetes mellitus is a diagnostic term for a group of disorders characterized by abnormal glucose homeostasis resulting in elevated blood sugar. There are many types of diabetes, but the two most common are type 1 (also referred to as insulin-dependent diabetes mellitus or IDDM) and type 2 (also referred to as non-insulin-dependent diabetes mellitus or NIDDM).
The etiology of the different types of diabetes is not the same; however, everyone with diabetes has two things in common: overproduction of glucose by the liver and little or no ability to move glucose out of the blood into the cells where it becomes the body’s primary fuel.
People who do not have diabetes rely on insulin, a hormone made in the pancreas, to move glucose from the blood into the cells of the body. However, people who have diabetes either do not produce insulin or can not efficiently use the insulin they produce; therefore, they can not move glucose into their cells. Glucose accumulates in the blood creating a condition called
hyperglycemia, and over time, can cause serious health problems. Diabetes is a syndrome with interrelated metabolic, vascular, and neuropathic components.
The metabolic syndrome, generally characterized by hyperglycemia, comprises alterations in carbohydrate, fat and protein metabolism caused by absent or markedly reduced insulin secretion and/or ineffective insulin action. The vascular syndrome consists of abnormalities in the blood vessels leading to cardiovascular, retinal and renal complications. Abnormalities in the peripheral and autonomic nervous systems are also part of the diabetic syndrome.
About 5% to 10% of the people who have diabetes have IDDM. These individuals do not produce insulin and therefore must inject insulin to keep their blood glucose levels normal. IDDM is characterized by low or undetectable levels of endogenous insulin production caused by destruction of the insulin-producing beta cells of the pancreas, the characteristic that most readily distinguishes IDDM from NIDDM. IDDM, once termed juvenile-onset diabetes, strikes young and older adults alike.
Approximately 90% to 95% of people with diabetes have NIDDM (type 2). NIDDM subjects produce insulin, but the cells in their bodies are insulin resistant: the cells do not respond properly to the hormone, so glucose accumulates in their blood. NIDDM is characterized by a relative disparity between endogenous insulin production and insulin requirements, leading to elevated blood glucose levels. In contrast to IDDM, there is always some endogenous insulin production in NIDDM; many NIDDM patients have normal or even elevated blood insulin levels, while other NIDDM patients have inadequate insulin production (Rotwein, R. et al. N. Engl. J. Med. 308, 65-71 (1983)). Most people diagnosed with NIDDM are age 30 or older, and half of all new cases are age 55 and older. Compared with whites and Asians, NIDDM is more common among Native Americans, African- Americans, Latinos, and Hispanics. In addition, the onset can be insidious or even clinically inapparent, making diagnosis difficult.
The primary pathogenic lesion on NIDDM has remained elusive. Many have suggested that primary insulin resistance of the peripheral tissues is the initial event. Genetic epidemiological studies have supported this view. Similarly, insulin secretion abnormalities have been argued as the primary defect in NIDDM. It is likely that both phenomena are important contributors to the disease process (Rimoin, D. L., et. al. Emery and Rimoin’s Principles and Practice of Medical Genetics 3ri Ed. 1: 1401-1402 (1996)).
Many people with NIDDM have sedentary lifestyles and are obese: they weigh approximately 20% more than the recommended weight for their height and build. Furthermore, obesity is characterized by hyperinsulinemia and insulin resistance, a feature shared with NIDDM, hypertension and atherosclerosis.
The patient with diabetes faces a 30% reduced lifespan. After age 45, people with diabetes are about three times more likely than people without diabetes to have significant heart disease and up to five times more likely to have a stroke. These findings emphasize the inter-relations between risks factors for NIDDM and coronary heart disease and the potential value of an integrated approach to the prevention of these conditions (Perry, I. J., et al., BMJ 310, 560-564 (1995)).
Diabetes has also been implicated in the development of kidney disease, eye diseases and nervous-system problems. Kidney disease, also called nephropathy, occurs when the kidney’s “filter mechanism” is damaged and protein leaks into urine in excessive amounts and eventually the kidney fails. Diabetes is also a leading cause of damage to the retina at the back of the eye and increases risk of cataracts and glaucoma. Finally, diabetes is associated with nerve damage, especially in the legs and feet, which interferes with the ability to sense pain and contributes to serious infections. Taken together, diabetes complications are one of the nation’s leading causes of death.
B. Obesity
Obesity and diabetes are among the most common human health problems in industrialized societies. In industrialized countries a third of the population is at least 20% overweight. In the United States, the percentage of obese people has increased from 25% at the end of the 1970’s, to 33% at the beginning the 1990’s. Obesity is one of the most important risk factors for NIDDM. Definitions of obesity differ, but in general, a subject weighing at least 20% more than the recommended weight for his/her height and build is considered obese. The risk of developing NIDDM is tripled in subjects 30% overweight, and three-quarters with NIDDM are overweight.
Obesity, which is the result of an imbalance between caloric intake and energy expenditure, is highly correlated with insulin resistance and diabetes in experimental animals and human.
However, the molecular mechanisms that are involved in obesity-diabetes syndromes are not clear. During early development of obesity, increased insulin secretion balances insulin resistance and protects patients from hyperglycemia (Le Stunff, et al. Diabetes 43, 696-702 (1989)). However, after several decades, β cell function deteriorates and non-insulin-dependent diabetes develops in about 20% of the obese population (Pederson, P. Diab. Metab. Rev. 5, 505-509 (1989)) and (Brancati, F. L., et al., Arch. Intern. Med. 159, 957-963 (1999)). Given its high prevalence in modern societies, obesity has thus become the leading risk factor for NIDDM (Hill, J. O., et al., Science 280, 1371-1374 (1998)). However, the factors which predispose a fraction of patients to alteration of insulin secretion in response to fat accumulation remain unknown.
Whether someone is classified as overweight or obese can be determined by a number of different methods, such as, on the basis of their body mass index (BMI) which is calculated by dividing body weight (kg) by height squared (m2). Thus, the units of BMI are kg/m2 and it is possible to calculate the BMI range associated with minimum mortality in each decade of life. Overweight is defined as a BMI in the range 25-30 kg/m2, and obesity as a BMI greater than 30 kg/m2(see table below). There are problems with this definition, such as, it does not take into account the proportion of body mass that is muscle in relation to fat (adipose tissue). To account for this, alternatively, obesity can be defined on the basis of body fat content: greater than 25% and 30% in males and females, respectively.
CLASSIFICATION OF WEIGHT BY BODY MASS INDEX (BMI)

As the BMI increases there is an increased risk of death from a variety of causes that is independent of other risk factors. The most common diseases associated with obesity are cardiovascular disease (particularly hypertension), diabetes (obesity aggravates the development of diabetes), gall bladder disease (particularly cancer) and diseases of reproduction. Research has shown that even a modest reduction in body weight can correspond to a significant reduction in the risk of developing coronary heart disease.
Obesity considerably increases the risk of developing cardiovascular diseases as well. Coronary insufficiency, atheromatous disease, and cardiac insufficiency are at the forefront of the cardiovascular complication induced by obesity. It is estimated that if the entire population had an ideal weight, the risk of coronary insufficiency would decrease by 25% and the risk of cardiac insufficiency and of cerebral vascular accidents by 35%. The incidence of coronary diseases is doubled in subjects less than 50 years of age who are 30% overweight.
C. Atherosclerosis
Atherosclerosis is a complex disease characterized by inflammation, lipid accumulation, cell death and fibrosis. Atherosclerosis is characterized by cholesterol deposition and monocyte infiltration into the subendothelial space, resulting in foam cell formation. Thrombosis subsequent to atherosclerosis leads to myocardial infarction and stroke. Atherosclerosis is the leading cause of mortality in many countries, including the United States. (See, e.g., Ruggeri, Nat Med (2002) 8: 1227-1234; Arehart et al, Circ Res, Circ. Res. (2008) 102:986-993.)
D. Osteoporosis
Osteoporosis is a disabling disease characterized by the loss of bone mass and microarchitectural deterioration of skeletal structure leading to compromised bone strength, which predisposes a patient to increased risk of fragility fractures. Osteoporosis affects more than 75 million people in Europe, Japan and the United States, and causes more than 2.3 million fractures in Europe and the United States alone. In the United States, osteoporosis affects at least 25% of all post-menopausal white women, and the proportion rises to 70% in women older than 80 years. One in three women older than 50 years will have an osteoporotic fracture that causes a considerable social and financial burden on society. The disease is not limited to women; older men also can be affected. By 2050, the worldwide incidence of hip fracture projected to increase by 310% in men and 240% in women. The combined lifetime risk for hip, forearm, and vertebral fractures presenting clinically is around 40%, equivalent to the risk for cardiovascular disease. Osteoporotic fractures therefore cause substantial mortality, morbidity, and economic cost. With an ageing population, the number of osteoporotic fractures and their costs will at least double in the next 50 years unless effective preventive strategies are developed. (See, e.g., Atik et al, Clin. Orthop. Relat. Res. (2006) 443: 19-24; Raisz, J. Clin. Invest. (2005) 115:3318-3325; and World Health Organization Technical Report Series 921 (2003), Prevention and
Management of Osteoporosis).
E. Inflammatory Bowel Disease (IBD)
Inflammatory bowel disease (IBD) is the general name for diseases that cause inflammation in the intestines and includes, e.g. Crohn’s disease, ulcerative colitis, and ulcerative proctitis. U.S. medical costs of inflammatory bowel disease for 1990 have been estimated to be $1.4 to $1.8 billion. Lost productivity has been estimated to have added an additional $0.4 to $0.8 billion, making the estimated cost of inflammatory bowel disease $1.8 to $2.6 billion. (See, e.g. , Pearson, Nursing Times (2004) 100:86-90; Hay et al., J. Clin.
Gastroenterol. (1992) 14:309-317; Keighley et al, Ailment Pharmacol. Ther. (2003) 18:66-70).
Enteritis refers to inflammation of the intestine, especially the small intestine, a general condition that can have any of numerous different causes. Enterocolitis refers to inflammation of the small intestine and colon.
Crohn’s disease (CD) is an inflammatory process that can affect any portion of the digestive tract, but is most commonly seen in the last part of the small intestine otherwise called the (terminal) ileum and cecum. Altogether this area is also known as the ileocecal region. Other cases may affect one or more of: the colon only, the small bowel only (duodenum, jejunum and/or ileum), the anus, stomach or esophagus. In contrast with ulcerative colitis, CD usually does not affect the rectum, but frequently affects the anus instead. The inflammation extends deep into the lining of the affected organ. The inflammation can cause pain and can make the intestines empty frequently, resulting in diarrhea. CD may also be called enteritis.
Granulomatous colitis is another name for CD that affects the colon. Ileitis is CD of the ileum which is the third part of the small intestine. Crohn’s colitis is CD affecting all or part of the colon.
Ulcerative colitis (UC) is an inflammatory disease of the large intestine, commonly called the colon. UC causes inflammation and ulceration of the inner lining of the colon and rectum. The inflammation of UC is usually most severe in the rectal area with severity diminishing (at a rate that varies from patient to patient) toward the cecum, where the large and small intestines join together. Inflammation of the rectum is called proctitis. Inflammation of the sigmoid colon (located just above the rectum) is called sigmoiditis. Inflammation involving the entire colon is termed pancolitis. The inflammation causes the colon to empty frequently resulting in diarrhea. As the lining of the colon is destroyed ulcers form releasing mucus, pus and blood. Ulcerative proctitis is a form of UC that affects only the rectum.
F. GPR119
GPR119 is a G protein-coupled receptor (GPR119; e.g. , human GPR119, GenBank® Accession No. AAP72125 and alleles thereof; e.g. , mouse GPR119, GenBank® Accession No. AY288423 and alleles thereof) and is selectively expressed on pancreatic beta cells. GPR119 activation leads to elevation of a level of intracellular cAMP, consistent with GPR119 being coupled to Gs. Agonists to GPR119 stimulate glucose-dependent insulin secretion in vitro and lower an elevated blood glucose level in vivo; see, e.g. , International Applications WO
04/065380 and WO 04/076413, and EP 1338651. In the literature, GPR119 has also been referred to as RUP3 (see, International Application WO 00/31258) and as Glucose-Dependent Insulinotropic Receptor GDIR (see, Jones, et. al. Expert Opin. Ther. Patents (2009), 19(10): 1339- 1359).
GPR119 agonists also stimulate the release of Glucose-dependent Insulinotropic Polypeptide (GIP), Glucagon-Like Peptide- 1 (GLP-1), and at least one other L-cell peptide, Peptide YY (PYY) (Jones, et. al. Expert Opin. Ther. Patents (2009), 19(10): 1339-1359); for specific references related to GPR119 agonists and the release of:
GIP, see Shah, Current Opinion in Drug Discovery & Development, (2009) 12:519-532; Jones, et al, Ann. Rep. Med. Chem. , (2009) 44: 149-170; WO 2007/120689; and WO 2007/120702;
GLP-1, see Shah, Current Opinion in Drug Discovery & Development, (2009) 12:519-532; Jones, et al, Ann. Rep. Med. Chem. , (2009) 44:149-170; Schwartz et. al., Cell Metabolism, 2010, 11 :445-447; and WO 2006/076231 ; and
PYY, see Schwartz et. al, Cell Metabolism, 2010, 11:445-447; and WO 2009/126245. As mentioned above, GPR119 agonists enhance incretin release and therefore can be used in treatment of disorders related to the incretins, such as, GIP, GLP-1, and PYY. However, a number of the incretins, such as, GIP and GLP-1, are substrates for the enzyme dipeptidyl peptidase-4 (DPP-IV). Jones and co-workers (Jones, et al, Ann. Rep. Med. Chem. , (2009) 44: 149- 170) have demonstrated that a combined administration of a GPR119 agonist, (2-Fluoro-4- methanesulfonyl-phenyl) – { 6-[4-(3 -isopropyl- [1,2,4] oxadiazol-5 -yl) -piperidin- 1 -yl] -5 -nitro- pyrimidin-4-yl} -amine (see, Compound B i l l in WO 2004/065380). and a DPP-IV inhibitor acutely increased plasma GLP-1 levels and improved glucose tolerance to a significantly greater degree than either agent alone.
G. Glucose-dependent Insulinotropic Polypeptide (GIP) Glucose-dependent insulinotropic polypeptide (GIP, also known as gastric inhibitory polypeptide) is a peptide incretin hormone of 42 amino acids that is released from duodenal endocrine K cells after meal ingestion. The amount of GIP released is largely dependent on the amount of glucose consumed. GIP has been shown to stimulate glucose-dependent insulin secretion in pancreatic beta cells. GIP mediates its actions through a specific G protein-coupled receptor, namely GIPR.
As GIP contains an alanine at position 2, it is an excellent substrate for DPP-IV, an enzyme regulating the degradation of GIP. Full-length GIP(l-42) is rapidly converted to bioinactive GIP(3-42) within minutes of secretion from the endocrine K cell. Inhibition of DPP- IV has been shown to augment GIP bioactivity. (See, e.g. , Drucker, Cell Metab (2006) 3: 153- 165; Mcintosh et al, Regul Pept (2005) 128: 159-165; Deacon, Regul Pept (2005) 128: 117-124; and Ahren et al., Endocrinology (2005) 146:2055-2059.). Analysis of full length bioactive GIP, for example in blood, can be carried out using N-terminal-specific assays (see, e.g. , Deacon et al., J Clin Endocrinol Metab (2000) 85:3575-3581).
Recently, GIP has been shown to promote bone formation. GIP has been shown to activate osteoblastic receptors, resulting in increases in collagen type I synthesis and alkaline phosphatase activity, both associated with bone formation. GIP has been shown to inhibit osteoclast activity and differentiation in vitro. GIP administration has been shown to prevent the bone loss due to ovariectomy. GIP receptor (GIPR) knockout mice evidence a decreased bone size, lower bone mass, altered bone microarchitecture and biochemical properties, and altered parameters for bone turnover, especially in bone formation. (See, e.g., Zhong et al., Am J Physiol Endocrinol Metab (2007) 292:E543-E548; Bollag et al., Endocrinology (2000) 141 : 1228-1235; Bollag et al., Mol Cell Endocrinol (2001) 177:35-41 ; Xie et al., Bone (2005) 37:759-769; and Tsukiyama et al., Mol Endocrinol (2006) 20: 1644-1651.)
The usefulness of GIP for maintaining or increasing bone density or formation has been acknowledged by the United States Patent and Trademark Office by issuance of United States Patent No. 6,410,508 for the treatment of reduced bone mineralization by administration of GIP peptide. However, current GIP peptide agonists suffer from a lack of oral bioavailability, negatively impacting patient compliance. An attractive alternative approach is to develop an orally active composition for increasing an endogenous level of GIP activity.
GPR119 agonists have been shown to stimulate the release of GIP; see Shah, Current Opinion in Drug Discovery & Development, (2009) 12:519-532; Jones, et al., Ann. Rep. Med. Chem., (2009) 44:149-170; WO 2007/120689; and WO 2007/120702.
H. Glucagon-Like Peptide-1 (GLP-1)
Glucagon-like peptide-1 (GLP-1) is an incretin hormone derived from the
posttranslational modification of proglucagon and secreted by gut endocrine cells. GLP-1 mediates its actions through a specific G protein-coupled receptor (GPCR), namely GLP-1R. GLP-1 is best characterized as a hormone that regulates glucose homeostasis. GLP-1 has been shown to stimulate glucose-dependent insulin secretion and to increase pancreatic beta cell mass. GLP-1 has also been shown to reduce the rate of gastric emptying and to promote satiety. The efficacy of GLP-1 peptide agonists in controlling blood glucose in type 2 diabetics has been demonstrated in several clinical studies [see, e.g. , Nauck et al., Drug News Perspect (2003) 16:413-422], as has its efficacy in reducing body mass [Zander et al., Lancet (2002) 359:824- 830].
GLP-1 receptor agonists are additionally useful in protecting against myocardial infarction and against cognitive and neurodegenerative disorders. GLP-1 has been shown to be cardioprotective in a rat model of myocardial infarction [Bose et al., Diabetes (2005) 54: 146- 151], and GLP-1 R has been shown in rodent models to be involved in learning andneuroprotection [During et al., Nat. Med. (2003) 9: 1173-1179; and Greig et al., Ann N Y Acad Sri (2004) 1035:290-315].
Certain disorders such as type 2 diabetes are characterized by a deficiency in GLP-1 [see, e.g., Nauck et al, Diabetes (2004) 53 Suppl 3:S190-196].
Current GLP-1 peptide agonists suffer from a lack of oral bioavailability, negatively impacting efficacy. Efforts to develop orally bioavailable non-peptidergic, small-molecule agonists of GLP-1 R have so far been unsuccessful (Mentlein, Expert Opin Investig Drugs (2005) 14:57-64). An attractive alternative approach is to develop an orally active composition for increasing an endogenous level of GLP-1 in the blood.
GPR119 agonists have been shown to stimulate the release of GLP-1, see Shah, Current Opinion in Drug Discovery & Development, (2009) 12:519-532; Jones, et al., Ann. Rep. Med. Chem., (2009) 44:149-170; Schwartz et. al., Cell Metabolism, 2010, 11:445-447; and WO2006/076231.
Clinical trials
Remogliflozin etabonate (RE) was shown to enhance urinary glucose excretion in rodents and humans. Early studies in diabetics improved plasma glucose levels.[3][4] Remogliflozin etabonate has been studied at doses up to 1000 mg.[5] A pair of 12-week phase 2b randomized clinical trials of diabetics published in 2015, found reductions in glycated hemoglobin and that it was generally well tolerated.[6]
Method of action
Remogliflozin etabonate is a pro-drug of remogliflozin. Remogliflozin inhibits the sodium-glucose transport proteins (SGLT), which are responsible for glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[7] Remogliflozin is selective for SGLT2.
Remogliflozin inhibits the sodium-glucose transport proteins, which are responsible for glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[3]
DPP IV inhibitors represent a novel class of agents that are being developed for the treatment or improvement in glycemic control in patients with type 2 diabetes. For example, DPP IV inhibitors and their uses are disclosed in WO 2002/068420, WO 2004/018467, WO 2004/018468, WO 2004/018469, WO 2004/041820, WO 2004/046148, WO 2005/051950, WO 2005/082906, WO 2005/063750, WO 2005/085246, WO 2006/027204, WO 2006/029769, WO2007/014886; WO 2004/050658, WO 2004/1 1 1051 , WO 2005/058901 , WO 2005/097798; WO 2006/068163, WO 2007/071738, WO 2008/017670; WO 2007/054201 or WO 2007/128761.

Chemical structures of remogliflozin etabonate (A), remogliflozin (B), sergliflozin (C), phlorizin (D), and T-1095 (E). Remogliflozin etabonate is metabolized to remogliflozin, its active form.
SYNTHESIS
JP 2011201871

CLIP, USE THE REF SHOWN BELOW
https://jstagebeta.jst.go.jp/article/cpb/64/7/64_c15-00982/_html
Volume 64 (2016) Issue 7 Pages 1009-1018,
Some glucopyranosyloxypyrazole derivatives such as 1a–d (Fig. 1) have been demonstrated to inhibit the low-affinity Na+-dependent glucose co-transporter SGLT2.1–3) Two types of SGLT are known, SGLT1 and SGLT2, both of which act as transmembrane glucose transporters. Although SGLT1 (high-affinity Na+-dependent glucose co-transporter) is expressed to some extent in the kidney and contributes to glucose reabsorption, it is mainly expressed in the small intestine, where it plays an important role in glucose absorption.4,5) SGLT2 is specifically expressed in the kidney and plays an important role in renal glucose reabsorption in the proximal tubule.6)Remogliflozin (1a), discovered at Kissei Pharmaceutical Co., Ltd., exhibits an inhibitory activity that is highly selective for SGLT2.7,8) Remogliflozin etabonate (1b), a prodrug of 1a, is metabolized to its active form 1a in the body, and may therefore be useful as a preventive or therapeutic agent for diseases attributable to hyperglycemia such as diabetes, complications related to diabetes, and obesity.9,10)
Remogliflozin etabonate (1b), a prodrug of 1a, is metabolized to its active form 1a in the body, and may therefore be useful as a preventive or therapeutic agent for diseases attributable to hyperglycemia such as diabetes, complications related to diabetes, and obesity.9,10)
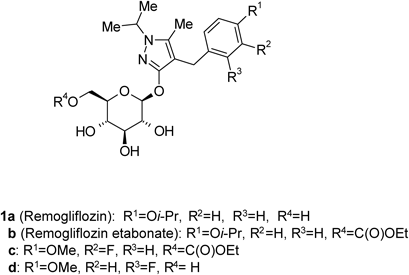
The synthetic strategy for 1b, given in Chart 1, shows that the 4-[(4-isopropoxyphenyl)methyl]-5-methyl-3-(2,3,4,6-tetra-O-acyl-β-D-glucopyranosyloxy)-1H-pyrazole derivative 2 is an important intermediate. O-Glycosylation of 4-[(4-isopropoxyphenyl)methyl]-5-methyl-1,2-dihydro-3H-pyrazol-3-one (3a) with a glycosyl donor (4 or 5) is therefore a key step in the production of 1b. Various O-glycosylation methods of 1,2-dihydro-3H-pyrazol-3-one derivatives 3have been reported, including the Koenigs–Knorr reaction, which employs a phase-transfer catalyst, and the Mitsunobu reaction.1,2,11–13) Although we also evaluated these methods for the preparation of 2, no successful results were obtained. Therefore, developing an efficient O-glycosylation method is strongly desired to establish scalable synthesis of 1b. We report here a convenient and practical method for the O-glycosylation of 3 with 2,3,4,6-tetra-O-acyl-α-D-glucopyranosyl bromide 5 via N1-acetylation of the pyrazole ring.
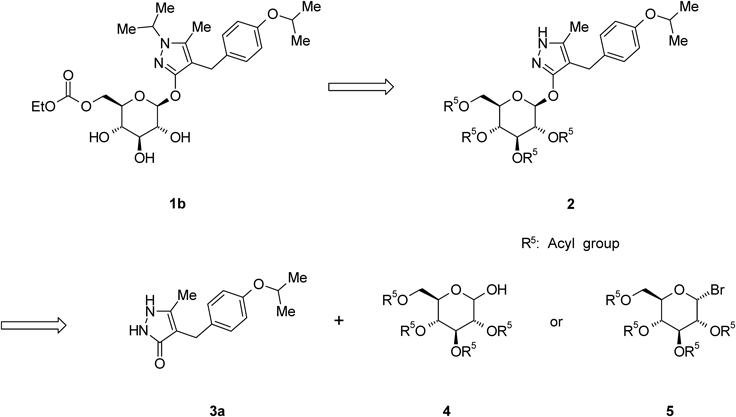
Remogliflozin etabonate (1b) was prepared by a four-step sequence starting from 2b, as shown in Chart 8. Introduction of an isopropyl group to 2b with 2-iodopropane in the presence of NaH in 1,3-dimethyl-2-imidazolidinone (DMI) provided 4-[(4-isopropoxyphenyl)methyl]-1-isopropyl-5-methyl-3-(2,3,4,6-tetra-O-acyl-β-D-glucopyranosyloxy)-1H-pyrazole derivative (15) in an 86% yield. The depivaloylation of 15 in the presence of sodium methoxide (MeONa) in MeOH provided Remogliflozin (1a) in a 99% yield. The reaction of 1a with ethyl chloroformate in the presence of 2,6-lutidine and pyridine in MeCN provided 16 as an ethanol solvate of 1b in a 72% yield. 16 was crystallized from a mixed solvent of methyl t-butyl ether (MTBE) and n-heptane to provide 1b in a 98% yield.
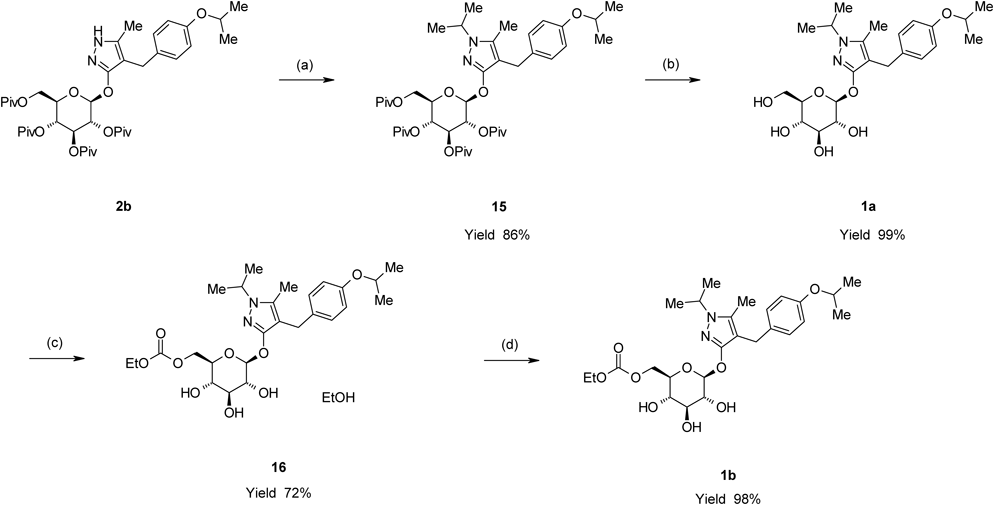
Chart 8. Preparation for 1b from 2bReagents: (a) 2-Iodopropane, NaH, DMI; (b) MeONa, MeOH; (c) Ethyl chloroformate, 2,3-lutidine, pyridine, MeCN; (d) MTBE, n-heptane.
In conclusion, an efficient and practical method for the synthesis of 2b, an important intermediate in the synthesis of 1b, was established. These results suggest that this O-glycosylation method could be applied in syntheses of additional glucopyranosyloxypyrazole derivatives exhibiting SGLT2 inhibitory activity such as 1c, d.
3-(β-D-Glucopyranosyloxy)-4-[(4-isopropoxyphenyl)methyl]-1-isopropyl-5-methyl-1H-pyrazole (1a)
A methanolic solution of 28% MeONa (1.93 g, 10 mmol) was added to a suspension of 15 (7.87 g, 10 mmol) in MeOH (75 mL) at room temperature. The mixture was then heated to 55°C and stirred for 3 h at this temperature. After cooling to 40°C, acetic acid (0.601 g, 10 mmol) was added dropwise to the reaction mixture. The reaction mixture was concentrated under reduced pressure to evaporate the methyl pivalate contained in the mixture. The residue was purified by silica gel chromatography (eluent dichloromethane–MeOH, 10 : 1) to provide 1a (4.45 g, 99% yield) as a pale yellowish oil.
[α]D20 −8.1 (c=1.0, DMSO).
IR (KBr) cm−1: 3407, 2975, 2931, 1506, 1466, 1384.
1H-NMR (CD3OD) δ: 1.26 (6H, d, J=6.0 Hz), 1.36 (6H, dd, J=3.8, 6.8 Hz), 2.09 (3H, s), 3.21–3.26 (1H, m), 3.33–3.43 (3H, m), 3.62–3.72 (3H, m), 3.77 (1H, dd, J=2.5, 12.1 Hz), 4.36–4.46 (1H, m), 4.46–4.55 (1H, m), 5.00–5.05 (1H, m), 6.76 (2H, d, J=8.7 Hz), 7.07 (2H, d, J=8.7 Hz).
13C-NMR (CD3OD) δ: 8.93 (q), 21.61 (q×2), 21.62 (q), 21.65 (q), 26.79 (t), 49.77 (d), 61.85 (t), 70.25 (d), 70.48 (d), 74.32 (d), 77.24 (d), 77.49 (d), 102.41 (d), 104.50 (s), 116.18 (d×2), 129.39 (d×2), 134.00 (s), 137.53 (s), 156.55 (s), 159.47 (s).
HR-MS (ESI) m/z: 451.2444 [M+H]+ (Calcd for C23H35N2O7: 451.2439).
5-Methyl-4-[4-(1-methylethoxy)benzyl]-1-(1-methylethyl)-1H-pyrazol-3-yl-6-O-(ethoxycarbonyl)-β-D-glucopyranoside (1b)
16 (1.50 g, 2.64 mmol) was dissolved in MTBE (10 mL) at 45°C The solution was concentrated under reduced pressure to evaporate EtOH. MTBE was added to the residue, and the weight was adjusted to 9.0 g. H2O (0.015 mL) and n-heptane (3.6 g) were added to the solution at 40°C and the solution was cooled to 25°C. The solution was seeded with 1a and stirred at 25°C for 3 h. The resulting slurry was warmed to 40°C, and then a mixed solvent of MTBE (0.44 g) and n-heptane (2.4 g) was added dropwise to the slurry while maintaining the temperature between 37 and 43°C. The slurry was stirred at 40°C for 1 h and for an additional 3 h at 10°C. The slurry was filtered and the wet cake washed successively with a mixed solvent of MTBE (1.5 g) and n-heptane (1.5 g) followed by n-heptane (3.0 g). The product was dried in vacuo at room temperature to give 1.35 g (98% yield) of
1a ( ERROR SHOULD BE 1b )as a white solid. mp 80–83°C. [α]D20 −19.3 (c=1.0, DMSO).
IR (KBr) cm−1: 3414, 2979, 1747, 1506, 1477, 1474, 1466, 1458, 1449, 1382, 1370, 1317.
1H-NMR (CD3OD) δ: 1.23 (3H, t, J=7.2 Hz), 1.26 (6H, d, J=6.1 Hz), 1.37 (6H, dd, J=2.3, 6.7 Hz), 2.07 (3H, s), 3.34–3.42 (4H, m), 3.61–3.69 (2H, m), 4.12 (2H, q, J=7.2 Hz), 4.21 (1H, dd, J=5.4, 11.5 Hz), 4.35 (1H, dd, J=1.7, 11.6 Hz), 4.35–4.45 (1H, m), 4.45–4.54 (1H, m), 5.04–5.06 (1H, m), 6.75 (2H, d, J=8.6 Hz), 7.06 (2H, d, J=8.6 Hz).
13C-NMR (CD3OD) δ: 9.70, 14.60, 22.43, 22.49, 22.54, 27.63, 50.53, 65.07, 67.67, 71.07, 71.21, 75.02, 75.56, 77.84, 103.25, 105.62, 116.98, 130.21, 134.81, 138.21, 156.65, 157.33, 159.99.
HR-MS (ESI) m/z: 523.2651 [M+H]+ (Calcd for C26H39N2O9: 523.2650).
5-Methyl-4-[4-(1-methylethoxy)benzyl]-1-(1-methylethyl)-1H-pyrazol-3-yl-6-O-(ethoxycarbonyl)-β-D-glucopyranoside Ethanolate (16)
A solution of ethyl chloroformate (522 mg, 4.81 mmol) in MeCN (1 mL) was added dropwise to a mixture of 1a(1.89 g, 4.19 mmol), 2,6-lutidine (672 mg, 6.28 mmol) and pyridine (13 mg, 0.17 mmol) in MeCN (10 mL) while maintaining the temperature between −3 and 3°C. The reaction mixture was stirred at 0°C for 2 h. After addition of glacial acetic acid (113 mg, 1.88 mmol), the reaction mixture was allowed to warm to room temperature. The reaction mixture was diluted with MTBE (10 mL) and 10% brine (5 mL), and then the layers were separated. The organic layer was washed twice with brine (5 mL), dried over anhydrous MgSO4 (2 g) and concentrated under reduced pressure. The residue was dissolved in EtOH (17 mL) and concentrated again under reduced pressure. EtOH was added to the residue, and the weight was adjusted to 9.3 g. To the EtOH solution, n-heptane (6 mL) was added and heated to 60°C to dissolve solids. The mixture was cooled to 45°C and stirred for 1 h at this temperature for an additional 1 h at 0–5°C. The slurry was filtered and the wet cake washed successively with a mixed solvent of EtOH (1.2 mL) and n-heptane (2.8 mL), which was cooled to 0°C, and then n-heptane (2.8 mL). The precipitate was dried in vacuo at room temperature to give 1.72 g (72% yield) of 16 as a white solid. mp 70–74°C. [α]D20 −17.7 (c=1.0, DMSO). IR (KBr) cm−1: 3353, 2980, 2926, 1753, 1731, 1508, 1477, 1467, 1449, 1386, 1371. 1H-NMR (CDCl3) δ: 1.23 (3H, t, J=7.0 Hz), 1.28 (3H, t, J=7.0 Hz), 1.30 (6H, d, J=6.0 Hz), 1.38 (6H, dd, J=2.3, 6.6 Hz), 2.06 (3H, s), 3.47–3.63 (6H, m), 3.71 (2H, q, J=7.0 Hz), 4.17 (2H, q, J=7.0 Hz), 4.24–4.31 (1H, m), 4.32–4.39 (2H, m), 4.43–4.52 (1H, m), 4.98 (1H, d, J=7.6 Hz), 6.77 (2H, d, J=8.6 Hz), 7.05 (2H, d, J=8.6 Hz). 13C-NMR (CDCl3) δ: 9.72, 14.21, 18.35, 22.09, 22.21, 22.25, 26.87, 49.44, 58.35, 64.23, 66.48, 69.49, 69.86, 73.65, 74.24, 76.44, 102.32, 104.67, 115.78, 129.10, 133.15, 136.55, 155.46, 155.96, 158.07. HR-MS (ESI) m/z: 523.2648 [M+H]+ (Calcd for C26H39N2O9: 523.2650).
1) Fujikura H., Fushimi N., Nishimura T., Nakabayashi T., Isaji M., PCT, WO 02/053573 (2002).
2) Ohsumi K., Umemura T., Matsueda H., Hatanaka T., Onuki A., Mae-zono K., Kageyama Y., Kondo N., PCT, WO 02/36602 (2002).
3) Kraemer G., Martin H., Adelgoss G., Dugi K., Duran A., Eickelmann P., Maier S., Pinnetti S., Ritter R., Schilcher G., Streicher R., Thomas L., PCT, WO 2007/080170 (2007).
4) Pajor A. M., Wright E. M., J. Biol. Chem., 267, 3557–3560 (1992).
5) Wright E. M., Am. J. Physiol. Renal Physiol., 280, F10–F18 (2001).
6) Kanai Y., Lee W. S., You G., Brown D., Hediger M. A., J. Clin. Invest., 93, 397–404 (1994).
7) Fujimori Y., Katsuno K., Nakashima I., Ishikawa-Takemura Y., Fujikura H., Isaji M., J. Pharmacol. Exp. Ther., 327, 268–276 (2008).
8) Mikhail N., Expert Opin. Investig. Drugs, 24, 1381–1387 (2015).
9) Isaji M., Curr. Opin. Investig. Drugs, 8, 285–292 (2007).
10) Katsuno K., Fujimori Y., Takemura Y., Hiratochi M., Itoh F., Komatsu Y., Fujikura H., Isaji M., J. Pharmacol. Exp. Ther., 320, 323–330 (2007).
11) Fujikura H., Nishimura T., Katsuno K., Hiratochi M., Iyobe A., Fujioka M., Isaji M., PCT, WO 01/16147 (2001).
12) Nishimura T., Fushimi N., Fujikura H., Katsuno K., Komatsu Y., Isaji M., PCT, WO 02/068439 (2002).
13) Washburn W. N., PCT, WO 03/020737 (2003).
14) Shng-Eun Y., Kyu Y. Y., Bull. Korean Chem. Soc., 10, 112 (1989).
17) Tokuoka Y., Kosobe T., Kawashima N., Kaji H., Nishino T., Ishizuka M., Kokai, Tokkyo Koho, JP2010053079 (2010).
PATENT
http://www.google.co.in/patents/WO2012040279A1?cl=en
In some embodiments, the pharmaceutical agent or the second pharmaceutical agent is a sulfonylurea selected from ethyl ((2R,35,45,5R,65)-3,4,5-trihydroxy-6-(4-(4-isopropoxybenzyl)- 1 -isopropyl-5-methyl- lH-pyrazol-3-yloxy)tetrahydro-2H-pyran-2-yl)methyl carbonate
(chemical structure shown below) and pharmaceutically acceptable salts, solvates, and hydrates thereof:

CLIP
Highly Selective Primary Alkoxycarboxylation and Esterification of Unprotected Pyranose Derivatives Mediated by Scandium(III) Triflate Catalysis
European Journal of Organic Chemistry (2012), 2012, (19), 3561-3565
http://onlinelibrary.wiley.com/doi/10.1002/ejoc.201200261/abstract

5-Methyl-1-(1-methylethyl)-4-({4-[(1-methylethyl)oxy]phenyl}methyl)-1H-pyrazol-3-yl 6-O- [(ethyloxy)carbonyl]--D-glucopyranoside (2): To a solution of 1 (1.5 Kg, 1.0 eq, 3.2 mol) in toluene (9.6 L) and ethanol (2.4 L) is added scandium triflate (2.4 g, 0.0015 eq) and diethylpyrocarbonate (597 g, 1.15 eq). The solution is heated to 45-55 ˚C for 1-6 hours before quenching with dilute acetic acid (4.5 L, 2.5 vol%). The mixture is cooled to 20 ˚C and the layers are allowed to separate. The bottom layer (aqueous) is discarded. The organic layer is washed again with dilute aq. acetic acid (4.5 L) and the aqueous layer discarded. The final organic layer is then concentrated under reduced pressure to about 2.25 volumes. MIBK (4.13 L), water (47 mL), and heptanes (12.8 L) are added and the desired compound is isolated by crystallization to afford a white solid. The cake is washed with 25% MIBK in heptanes and then dried under reduced pressure (30 ˚C) to afford the title compound 2 as a white solid (1.55 kg, 92% yield).
FTIR (ATR) Vmax cm-1: 3225, 2978, 1744, 1610, 1504, 1382, 1295, 1079, 1052; 1H NMR (500 MHz, DMSO-d6) 1.17 (t, J = 7.1 Hz, 3H), 1.22 (d, J = 6.1 Hz, 6H), 1.27 (dd, J1 = 6.7 Hz, J2 = 8.3 Hz, 6H), 2.06 (s, 3H), 3.12-3.29 (m, 3H), 3.38 (ddd, J1 = 1.8 Hz, J2 =6.1 Hz, J3 = 10.0 Hz, 1H), 3.51 (s, 2H), 4.08 (q, J = 7.1 Hz, 2H), 4.10 (dd, J1 = 6.1 Hz, J2 = 11.7 Hz, 1H), 4.29 (dd, J1 = 1.8 Hz, J2 = 11.7 Hz, 1H), 4.34 (sp, J = 6.4 Hz, 1H), 4.50 (sp, J = 6.0 Hz, 1H), 5.12 (d, J = 7.9 Hz, 1H), 5.14 (d, J = 5.3 Hz, 1H), 5.25 (d, J = 5.8 Hz, 1H), 5.32 (d, J = 5.4 Hz, 1H), 6.75 (d, J = 8.6 Hz, 2H), 7.08 (d, J = 8.6 Hz, 2H);
13C NMR (125 MHz, DMSO-d6) 9.1, 13.9, 21.8, 21.9, 22.2, 26.2, 48.3, 63.4, 66.6, 68.9, 69.5, 73.2, 73.8, 76.3, 100.6, 102.8, 115.3, 129.0, 133.2, 135.5, 154.4, 155.3, 157.8 ppm;
HRMS (+ESI): [M+H]+ calcd.: 523.2656, found: 523.2654.
PATENT
US 7,084,123
http://www.google.co.in/patents/US7084123
EXAMPLE 1
3-(6-O-Ethoxycarbonyl-β-D-glucopyranosyloxy)-4-[(4-isopropoxyphenyl)methyl]-1-isopropyl-5-methylpyrazole
To a solution of 3-(β-D-glucopyranosyloxy)-4-[(4-isopropoxyphenyl)methyl]-1-isopropyl-5-methylpyrazole (0.10 g) in 2,4,6-trimethylpyridine (1 mL) was added ethyl chloroformate (0.072 g), and the mixture was stirred at room temperature overnight. To the reaction mixture were added citric acid monohydrate (3.3 g) and water, and the resulting mixture was purified by ODS solid phase extraction (washing solvent: distilled water, eluent: methanol). Further purification by column chromatography on silica gel (eluent: dichloromethane/methanol=1011) and recrystalization (recrystalization solvent: ethyl acetate/hexane=1/3) afforded 3-(6-O-ethoxycarbonyl-β-D-glucopyranosyloxy)-4-[(4-isopropoxyphenyl)methyl]-1-isopropyl-5-methylpyrazole (0.084 g).
1H-NMR (CD3OD) δ ppm: 1.23 (3H, t, J=7.0 Hz), 1.26 (6H, d, J=5.8 Hz), 1.30–1.40 (6H, m), 2.07 (3H, s), 3.25–3.45 (4H, m), 3.60–3.70 (2H, m), 4.12 (2H, q, J=7.0 Hz), 4.21 (1H, dd, J=5.4, 11.6 Hz), 4.34 (1H, dd, J=1.7, 11.6 Hz), 4.35–4.45 (1H, m), 4.45–4.55 (1H, m), 5.00–5.10 (1H, m), 6.70–6.80 (2H, m), 7.00–7.10 (2H, m)
CLIP
References
- Statement on a nonproprietory name adopted by the USAN council
- Yahoo Finance http://finance.yahoo.com/news/avolynt-announces-completion-phase-2b-143600142.html. Missing or empty
|title=(help) - “Remogliflozin etabonate, a selective inhibitor of the sodium-glucose transporter 2, improves serum glucose profiles in type 1 diabetes.”. 35. Nov 2012: 2198–200. doi:10.2337/dc12-0508.PMID 23011728.
- “Remogliflozin etabonate, a selective inhibitor of the sodium-dependent transporter 2 reduces serum glucose in type 2 diabetes mellitus patients.”. 14. Jan 2012: 15–22. doi:10.1111/j.1463-1326.2011.01462.x. PMID 21733056.
- “Randomized trial showing efficacy and safety of twice-daily remogliflozin etabonate for the treatment of type 2 diabetes.”. Diabetes Obes Metab. 17: 94–7. Jan 2015.doi:10.1111/dom.12391. PMID 25223369.
- “Randomized efficacy and safety trial of once-daily remogliflozin etabonate for the treatment of type 2 diabetes.”. 17. Jan 2015: 98–101. doi:10.1111/dom.12393. PMID 25238025.
- Prous Science: Molecule of the Month November 2007 Archived January 6, 2008, at the Wayback Machine.
REFERENCES
1: Nakano S, Katsuno K, Isaji M, Nagasawa T, Buehrer B, Walker S, Wilkison WO, Cheatham B. Remogliflozin Etabonate Improves Fatty Liver Disease in Diet-Induced Obese Male Mice. J Clin Exp Hepatol. 2015 Sep;5(3):190-8. doi: 10.1016/j.jceh.2015.02.005. Epub 2015 Apr 28. PubMed PMID: 26628836; PubMed Central PMCID: PMC4632078.
2: Mikhail N. Remogliflozin etabonate: a novel SGLT2 inhibitor for treatment of diabetes mellitus. Expert Opin Investig Drugs. 2015;24(10):1381-7. doi: 10.1517/13543784.2015.1061501. Epub 2015 Aug 14. PubMed PMID: 26288025.
3: Mikhail N. Remogliflozin etabonate : a novel SGLT2 inhibitor for treatment of diabetes mellitus. Expert Opin Investig Drugs. 2015 Aug 14:1-7. [Epub ahead of print] PubMed PMID: 26271274.
4: O’Connor-Semmes R, Walker S, Kapur A, Hussey EK, Ye J, Wang-Smith L, Tao W, Dobbins RL, Cheatham B, Wilkison WO. Pharmacokinetics and Pharmacodynamics of the SGLT2 Inhibitor Remogliflozin Etabonate in Subjects with Mild and Moderate Renal Impairment. Drug Metab Dispos. 2015 Jul;43(7):1077-83. doi: 10.1124/dmd.114.062828. Epub 2015 May 1. PubMed PMID: 25934577.
5: Sykes AP, Kemp GL, Dobbins R, O’Connor-Semmes R, Almond SR, Wilkison WO, Walker S, Kler L. Randomized efficacy and safety trial of once-daily remogliflozin etabonate for the treatment of type 2 diabetes. Diabetes Obes Metab. 2015 Jan;17(1):98-101. doi: 10.1111/dom.12393. Epub 2014 Nov 3. PubMed PMID: 25238025.
6: Sykes AP, O’Connor-Semmes R, Dobbins R, Dorey DJ, Lorimer JD, Walker S, Wilkison WO, Kler L. Randomized trial showing efficacy and safety of twice-daily remogliflozin etabonate for the treatment of type 2 diabetes. Diabetes Obes Metab. 2015 Jan;17(1):94-7. doi: 10.1111/dom.12391. Epub 2014 Nov 3. PubMed PMID: 25223369.
7: Jackson VM, Price DA, Carpino PA. Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies. Expert Opin Investig Drugs. 2014 Aug;23(8):1055-66. doi: 10.1517/13543784.2014.918952. Epub 2014 Jul 7. Review. PubMed PMID: 25000213.
8: Kapur A, O’Connor-Semmes R, Hussey EK, Dobbins RL, Tao W, Hompesch M, Smith GA, Polli JW, James CD Jr, Mikoshiba I, Nunez DJ. First human dose-escalation study with remogliflozin etabonate, a selective inhibitor of the sodium-glucose transporter 2 (SGLT2), in healthy subjects and in subjects with type 2 diabetes mellitus. BMC Pharmacol Toxicol. 2013 May 13;14:26. doi: 10.1186/2050-6511-14-26. PubMed PMID: 23668634; PubMed Central PMCID: PMC3700763.
9: Hussey EK, Kapur A, O’Connor-Semmes R, Tao W, Rafferty B, Polli JW, James CD Jr, Dobbins RL. Safety, pharmacokinetics and pharmacodynamics of remogliflozin etabonate, a novel SGLT2 inhibitor, and metformin when co-administered in subjects with type 2 diabetes mellitus. BMC Pharmacol Toxicol. 2013 Apr 30;14:25. doi: 10.1186/2050-6511-14-25. PubMed PMID: 23631443; PubMed Central PMCID: PMC3682882.
10: O’Connor-Semmes RL, Sandefer EP, Hussey EK, Tao W, Doll WJ, Page RC, Dobbins R. Regional gastrointestinal delivery of remogliflozin etabonate in humans. Biopharm Drug Dispos. 2013 Mar;34(2):79-86. doi: 10.1002/bdd.1824. Epub 2013 Jan 7. PubMed PMID: 23111980.
11: Mudaliar S, Armstrong DA, Mavian AA, O’Connor-Semmes R, Mydlow PK, Ye J, Hussey EK, Nunez DJ, Henry RR, Dobbins RL. Remogliflozin etabonate, a selective inhibitor of the sodium-glucose transporter 2, improves serum glucose profiles in type 1 diabetes. Diabetes Care. 2012 Nov;35(11):2198-200. doi: 10.2337/dc12-0508. Epub 2012 Sep 25. PubMed PMID: 23011728; PubMed Central PMCID: PMC3476920.
12: Sigafoos JF, Bowers GD, Castellino S, Culp AG, Wagner DS, Reese MJ, Humphreys JE, Hussey EK, O’Connor Semmes RL, Kapur A, Tao W, Dobbins RL, Polli JW. Assessment of the drug interaction risk for remogliflozin etabonate, a sodium-dependent glucose cotransporter-2 inhibitor: evidence from in vitro, human mass balance, and ketoconazole interaction studies. Drug Metab Dispos. 2012 Nov;40(11):2090-101. doi: 10.1124/dmd.112.047258. Epub 2012 Jul 30. PubMed PMID: 22851617.
13: Papazafiropoulou AK, Kardara MS, Pappas SI. Challenges for the treatment of diabetes mellitus. Recent Pat Endocr Metab Immune Drug Discov. 2011 Sep;5(3):203-9. Review. PubMed PMID: 21913881.
14: Dobbins RL, O’Connor-Semmes R, Kapur A, Kapitza C, Golor G, Mikoshiba I, Tao W, Hussey EK. Remogliflozin etabonate, a selective inhibitor of the sodium-dependent transporter 2 reduces serum glucose in type 2 diabetes mellitus patients. Diabetes Obes Metab. 2012 Jan;14(1):15-22. doi: 10.1111/j.1463-1326.2011.01462.x. Epub 2011 Oct 30. PubMed PMID: 21733056.
15: Fujimori Y, Katsuno K, Nakashima I, Ishikawa-Takemura Y, Fujikura H, Isaji M. Remogliflozin etabonate, in a novel category of selective low-affinity sodium glucose cotransporter (SGLT2) inhibitors, exhibits antidiabetic efficacy in rodent models. J Pharmacol Exp Ther. 2008 Oct;327(1):268-76. doi: 10.1124/jpet.108.140210. Epub 2008 Jun 26. PubMed PMID: 18583547.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
5-methyl-4-[4-(1-methylethoxy)benzyl]-1-(1-methylethyl)-1H-pyrazol-3-yl 6-O-(ethoxycarbonyl)-β-D-glucopyranoside
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | 442201-24-3 |
| ATC code | none |
| ChemSpider | 8047110 |
| UNII | TR0QT6QSUL |
| KEGG | D10055 |
| ChEMBL | CHEMBL494323 |
| Chemical data | |
| Formula | C26H38N2O9 |
| Molar mass | 522.586 g/mol |
REMOGLIFLOZIN AND REMOGLIFLOZIN ETABONATE




Chemical Information
| M.Wt | Formula | CAS No. | Synonyms |
|---|---|---|---|
| 450.53 | C23H34N2O7 | 329045-45-6 | Remogliflozin A; (2R,3S,4S,5R,6S)-2-(hydroxymethyl)-6-((4-(4-isopropoxybenzyl)-1-isopropyl-5-methyl-1H-pyrazol-3-yl)oxy)tetrahydro-2H-pyran-3,4,5-triolBMS-790052; EBP 883; BMS 790052 |
Structure Information of Remogliflozin
| Smiles | O[C@H]([C@H]([C@@H]([C@@H](CO)O1)O)O)[C@@H]1OC2=NN(C(C)C)C(C)=C2CC3=CC=C(OC(C)C)C=C3 |
|---|
///////////GSK-189075; GSK-189075A, GSK189075A, GSK-189075A, GSK 189075, GSK18907, GSK-18907, GSK 18907, Remogliflozin, O-glycosylation , 1,2-dihydro-3H-pyrazol-3-ones derivative , N1-acetylation , glucopyranosyloxypyrazole derivative , Remogliflozin etabonate, 442201-24-3, 189075, BHV-091009, GSK-189075, GSK-189075A, KGT-1681, BHV Pharma Kissei , GlaxoSmithKline, SGLT-2 antagonist, type 2 diabetes, non-alcoholic steatohepatitis, (NASH), obesity,
OC1C(COC(=O)OCC)OC(C(O)C1O)Oc(nn(C(C)C)c2C)c2Cc3ccc(cc3)OC(C)C