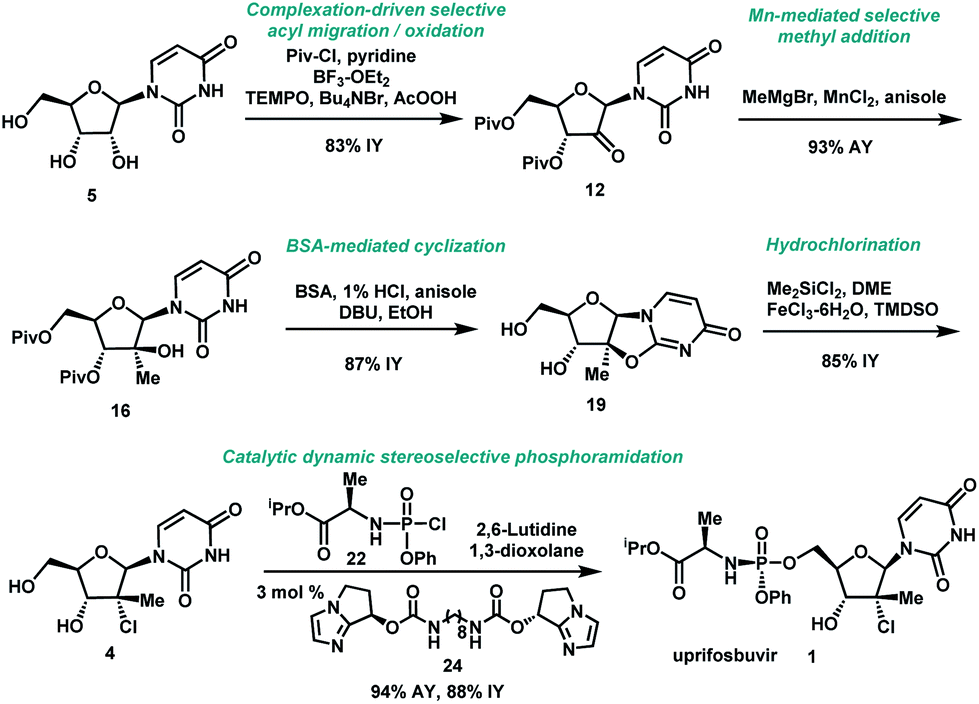
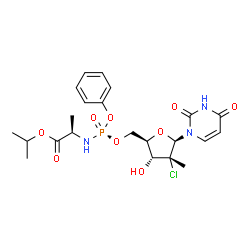
Uprifosbuvir
MK 3682, IDX 21437
ウプリホスブビル;
| Formula |
C22H29ClN3O9P
|
| CAS |
1496551-77-9
|
| Mol weight |
545.9071
|
уприфосбувир [Russian] [INN]
أوبريفوسبوفير [Arabic] [INN]
propan-2-yl (2R)-2-[[[(2R,3R,4R,5R)-4-chloro-5-(2,4-dioxopyrimidin-1-yl)-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate
Isopropyl (2R)-2-{[(R)-{[(2R,3R,4R,5R)-4-chloro-5-(2,4-dioxo-3,4-dihydro-1(2H)-pyrimidinyl)-3-hydroxy-4-methyltetrahydro-2-furanyl]methoxy}(phenoxy)phosphoryl]amino}propanoate
IDX-21437, DB15206, SB18784, D10996, Q27281714
Uprifosbuvir (MK-3682) is an antiviral drug developed for the treatment of Hepatitis C. It is a nucleotide analogue which acts as an NS5B RNA polymerase inhibitor. It is currently in Phase III human clinical trials.[1][2][3]
Uprifosbuvir is under investigation in clinical trial NCT02332707 (Efficacy and Safety of Grazoprevir (MK-5172) and Uprifosbuvir (MK-3682) With Elbasvir (MK-8742) or Ruzasvir (MK-8408) for Chronic Hepatitis C Genotype (GT)1 and GT2 Infection (MK-3682-011)).
Hepatitis C viruss (HCV) have the newly-increased patients of 3-4 million every year, and World Health Organization (WHO) is estimated in global sense More than 200,000,000, in China more than 10,000,000 patients, HCV belongs to flaviviridae hepatovirus virus to dye person.Long-term hepatitis C virus Gently to inflammation, weight is to liver cirrhosis, hepatocarcinoma for poison infection.And during hepatitis C cirrhosis patients in decompensation, can there are various complication, such as abdomen Water abdominal cavity infection, upper gastrointestinal hemorrhage, hepatic encephalopathy, hepatorenal syndrome, liver failure etc. are showed.The side of HCV infection is treated initially Method is interferon and interferon and ribavirin combination therapy, and only 50% therapist has reaction, and interferon to the method With obvious side effect, such as flu-like symptoms, body weight lower and fatigue and weak, and interferon and ribavirin Conjoint therapy then produces sizable side effect, including haemolysis, anemia and tired etc..
U.S. FDA have approved multiple HCV medicines, including the polymerization of protease inhibitor, ucleosides and non-nucleoside in recent years Enzyme inhibitor and NS5A inhibitor etc..The protease inhibitor class medicine of FDA approvals has three:VX‐950 (Telaprevir), SCH-503034 (Boceprevir) and TMC435 (Simeprevir), the shortcoming of protease inhibitor is It is also easy to produce that mutation, toxicity is big, poor bioavailability, it is effective to individual other gene type.Eggs of the Telaprevir as the first generation White enzyme inhibitor has logged out market.The second filial generation and third generation protease inhibitor of high activity and wide spectrum is mainly used as and other One of component of drug combination of hepatitis C medicine.
NS5A inhibitor is the highly active anti-HCV medicament of a class.The most representative Daclatasive for having BMS, The Ombitasvir of the Ledipasvir and AbbVie of Gilead, as this kind of medicine independent medication is easy to produce drug resistance, They treat one of drug component of HCV primarily as drug combination.
The AG14361 of hepatitis C is generally divided into two kinds of ucleosides and non-nucleoside.At present, clinically only Suo Feibu One ucleosides hepatitis C medicine of Wei is listed by FDA approvals, and other are still in the anti-hepatitis C virus medicine of ucleosides of clinical experimental stage Thing also has the MK-3682 (IDX21437) of Mo Shadong, the AL-335 of the ACH-3422 and Alios of Achillion drugmakers.Third Hepatitis virus have the features such as Multi-genotype and fast variation, and single medicine treatment hepatitis C has generation drug resistance fast, to part Genotype cure rate is low and the various defects such as course for the treatment of length.In order to overcome these defects, the treatment of drug combination is primarily now taken Scheme, in order to overcome these defects, primarily now takes the therapeutic scheme of drug combination, the Sovaldi conducts of FDA approval listings The key component of drug combination, for the patient of 4 type of 1 type of gene and gene be Suo Feibuwei, profit Ba Wei woodss and Polyethylene Glycol-α- The drug combination of interferon three, the course for the treatment of are 12 weeks;For 1 type of gene and the patient of 3 types, the big woods joints of Suo Feibuwei and Li Ba Medication, the course for the treatment of are respectively 12 weeks and 24 weeks.- 2016 years 2013, FDA ratified Suo Feibuwei and NS3 protein inhibitors again in succession Simeprevir shares the patient of 1 type of therapeutic gene;The NS5A inhibitor Daclatavir therapeutic genes 1 of Suo Feibuwei and BMS With the patient of 3 types.Harvoni is the patient that Suo Feibuweijia NS5A inhibitor Ledipasvir is used for 1 type of gene.Even if using Same nucleoside, the NS5A inhibitor and/or NS3 protease inhibitor for sharing varying strength can effectively extend composition of medicine Clinical application range and Shorten the Treatment Process.In June, 2016, FDA have approved Suo Feibuwei and more potent secondary NS5A inhibitor Velpatasvir shares the hepatitis C patient suitable for all gene types, it is not necessary to carry out genetic test.Just in three phases clinic Suo Feibuwei, NS5A inhibitor Velpatasvir and NS3 protease inhibitor Voxilaprevir goes for all of disease People, is try to the course for the treatment of and shortened to 8 weeks from 12 weeks.Suo Feibuwei just in clinical trial target spots different with hepatitis C virus are directed to Drug regimen (such as Suo Feibuweijia new type NS 5A inhibitor Velpatasvir and/or protease inhibitor GS5816), its knot Fruit show than single drug more wide spectrum, effectively, and can be with Shorten the Treatment Process.MSD Corp. is by MK-3682 and NS5A inhibitor Grazoprevir and/or protease inhibitor Elbasvir is used as new drug regimen, effective for all genotype of HCV, And further shorten to the course for the treatment of of 8 weeks.New deuterated nucleoside phosphoric acid ester compound disclosed in patent of the present invention, especially The double deuterated compound such as VI-1b2 in 5 ‘-position, shows than the more preferable bioavailability of former compound MK-3682 and longer partly declines Phase.In addition, this kind of novel nucleoside phosphoramidate is significantly superior to the Suo Feibuwei of clinical practice in terms of anti-hepatitis C activity, On sugared ring, chlorine atom replaces fluorine atom, and cytotoxicity is significantly reduced in surveyed cell line.By to base, sugared ring With the transformation and optimization of prodrug moiety system, the anti-hepatitis C activity of partial synthesis compound is higher than Suo Feibuwei 2-10 times, meanwhile, In the optimization of metabolism key position, synthesis compound shows that in blood plasma the higher metabolic stabilities of peso Fei Buwei and chemistry are steady It is qualitative.Therefore this kind of new deuterated nucleotide phosphate and NS5A inhibitor and/or egg as shown in formula a, a1, a2, b, b1, b2 The newtype drug combination constituted by white enzyme inhibitor is with extremely wide application prospect.
Deuterium is the naturally occurring hydrogen isotope of nature, the deuterated isotopic body in common drug all containing trace.Deuterium without It is malicious, “dead”, it is safe to human body, C-D keys are more stable (6-9 times) than c h bond, hydrogen is replaced with after deuterium, can extend medicine Half-life, while pharmacologically active (shape difference of H and D is little, J Med Chem.2011,54,2529-2591) is not affected, in addition Deuterated medicine usually shows more preferable bioavailability and less toxicity, and the active ribonucleoside triphosphote of its metabolism is more stable, So deuterated nucleoside phosphoramidate will be better than corresponding nucleoside medicine in the curative effect of clinical practice.For example, 2013 It is exactly a deuterated compound that the nucleoside anti hepatitis C virus drug ACH-3422 of clinical trial is in the approval of year FDA, with non-deuterium (WO2014169278, WO are 2014169280) than having higher bioavailability and longer half-life for the former compound phase in generation.
Based on above-mentioned present Research, we design and are prepared for the new deuterated nucleoside that compound VI-1b2 is representative Phosphoramidate.Below we will be described in the architectural feature of deuterated nucleoside phosphoramidate of our inventions, preparation method, Antiviral activity experimental result and it as anti-hepatitis c virus drug combination key component and NS5A inhibitor and/ Or the drug regimen of protease inhibitor is in the application of anti-virus aspect.
The EPA awarded the greener reaction conditions to the pharmaceutical company Merck & Co. for building a prodrug synthesis that eliminated the use of toxic reagents. Prodrugs are molecules that get metabolized by our bodies into an active pharmaceutical. Some hepatitis C and HIV medications are prodrugs and get synthesized through a method call pronucleotide (ProTide) synthesis. The method uses toxic and corrosive thionyl chloride, plus an excess of expensive pentafluorophenol that generates a lot of waste. Merck’s new method creates their target compounds in 90 to 92% yields without these reagents and eliminates the need for halogenated solvents entirely through strategic catalyst loading and the use of different starting materials from the traditional route.
The design of greener chemicals award went to the development of more environmentally friendly versions of chemicals called thermoset binders, which can serve as carpet adhesives and are involved in the manufacture of mineral and fiberglass products. Generally, these chemicals are based on formaldehyde or polycarboxylic acids, and they can give off toxic formaldehyde and often use small amounts of sulfuric and hypophosphorous acid as catalysts to activate them. The insulation and commercial roofing company Johns Manville created a new binder based on the reaction between renewable dextrose, fructose, and other simple sugars, bound together by the α-carbon-containing cross-linking agent glyoxal. The reaction also uses a biodegradable acid in water as a catalyst. The binder can be made in just one step instead of the traditional multistep synthesis. Also, the synthesis can be done directly at the manufacturing site, instead of beforehand like with the traditional approach, meaning this new binder creates fewer of the health and environmental hazards that come from storage and transportation.
SYN
US 20170226146,
Paper
Organic Process Research & Development (2021), 25(3), 661-667.
https://pubs.acs.org/doi/10.1021/acs.oprd.0c00487

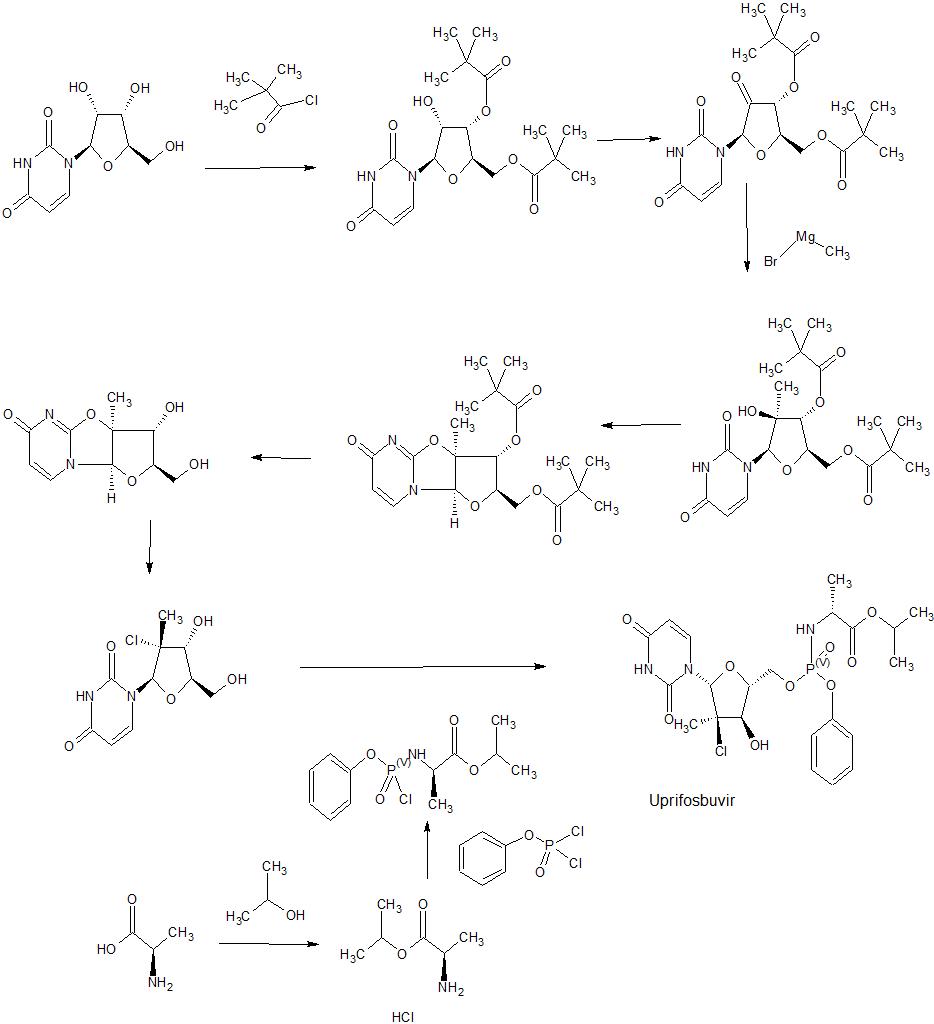
A novel application of the synthesis of pronucleotide (ProTide) 5′-phosphoramidate monoesters promoted by aluminum-based Lewis acids is described. In the multikilogram synthesis of uprifosbuvir (MK-3682, 1), a clinical candidate for the treatment of hepatitis C, this methodology provided >100:1 diastereoselectivity at the phosphorus stereocenter and >100:1 selectivity for the 5′-mono phosphorylation over undesired bisphosphorylation side products. The high diastereoselectivity and mono/bis ratio achieved enabled elimination of the tedious workup associated with the tert-butyl magnesium chloride protocol commonly used to install this functionality in similar nucleotide prodrugs, achieving a near doubling of the isolated yield from 45% to 81%. The process development and purity control strategy of MK-3682, as well as handling of the pyrophoric reagent on scale, will also be discussed.
PAPER
Science (Washington, DC, United States) (2020), 369(6504), 725-730.
Science (Washington, DC, United States) (2017), 356(6336), 426-430.
Chemical Science (2017), 8(4), 2804-2810.
PATENT
CN 106543253
https://patents.google.com/patent/CN106543253A/zh
PATENT
WO 2014058801
https://patents.google.com/patent/WO2014058801A1/en
Example 1
Preparation of 2′-Chloro Nucleoside Analogs
Ethyl (3R)-2-chloro-3-[(4R)-2,2-dimethyl-l,3-dioxolan-4-yl]-3-hydroxy-2- methylpropanoate (A2):
[00273] A 5 L flange flask was fitted with a thermometer, nitrogen inlet, pressure equalizing dropping funnel, bubbler, and a suba»seal. Methyl lithium solution (1.06 L, 1.6 M in diethylether, 1.7 equiv.) was added, and the solution was cooled to about -25 °C.
Diisopropyl amine (238 ml, 1.7 equiv.) was added using the dropping funnel over about 40 minutes. The reaction was left stirring, allowing to warm to ambient temperature overnight. C02(s)/acetone cooling was applied to the LDA solution, cooling to about -70 °C.
[00274] i?-Glyceraldehyde dimethylacetal solution (50% in DCM) was evaporated down to -100 mbar at a bath temp of 35 °C, to remove the DCM, then azeotroped with anhydrous hexane (200 ml), under the same Buchi conditions. 1H NMR was used to confirm that all but a trace of DCM remained.
[00275] The fresh aldehyde (130 g, 1 mol) and ethyl 2-chloropropionionate (191 ml, 1.5 equiv.) were placed in a 1 L round bottom flask, which was filled with toluene (800 ml). This solution was cooled in a C02(s)/acetone bath, and added via cannula to the LDA solution over about 50 minutes, keeping the internal temperature of the reaction mixture cooler than -60 °C. The mixture was stirred with cooling (internal temp, slowly fell to ~ -72 °C) for 90 min, then warmed to room temperature over 30 minutes using a water bath. This solution was added to a sodium dihydrogen phosphate solution equivalent to 360 g of NaH2P04 in 1.5 L of ice/water, over about 10 minutes, with ice-bath cooling. The mixture was stirred for 20 minutes, then transferred to a sep. funnel, and partitioned. The aqueous layer was further extracted with EtOAc (2 x 1 L), and the combined organic extracts were dried over sodium sulfate. The volatiles were removed in vacuo (down to 20 mbar). The resultant oil was hydrolyzed crude.
(3R,4R,5R)-3-chIoro-4-hydroxy-5-(hydroxymethyI)-3-methyIoxoIan-2-one (A4):
H O CI
[00276] The crude oil A2 was taken up in acetic acid (1.5 L, 66% in water) and heated to 90 °C over one hour, then at held at that temperature for one hour. Once the mixture had cooled to room temperature, the volatiles were removed in vacuo, and azeotroped with toluene (500 ml). The resultant oil was combined with some mixed material from an earlier synthesis and columned in two portions (each -1.25 L of silica, 38→ 75% EtOAc in DCM). The lower of the two main spots is the desired material; fractions containing this material as the major component were combined and the solvent removed in vacuo to give 82 g of orange solid whose 1 H NMR showed the material to be of about 57% purity (of the remainder 29% was the indicated epimer). This material was recrystallized from
toluene/butanone (600 ml / -185 ml), the butanone being the ‘good’ solvent. The resultant solid was filtered washing with toluene and hexane, and dried in vacuo to give product of about 92% purity (30 g).
(2R,3R,4R)-2-[(benzoyIoxy)methyI]-4-chIoro-4-methyI-5-oxooxoIan-3-yI benzoate
(A5):
[00277] A 2 L 3 -neck round bottom flask was fitted with an overhead stirrer, thermometer and pressure equalizing dropping funnel (→N2). The intermediate A4 (160 mmol) in acetonitrile (1 L) was added, followed by 4-dimethylaminopyridine (3.2 mmol) and benzoyl chloride (352 mmol). Finally triethylamine (384 mmol) was added over 10 minutes using the dropping funnel. The addition of the triethylamine is accompanied by a mild exotherm, which obviated the addition of a cold water bath to keep the internal temperature below 25 °C. The reaction was stirred at ambient temperature for 2.5 hours. The reaction mixture was transferred to a sep. funnel with EtOAc (2 L) and half saturated brine (2 L), and partitioned. The aqueous layer was re-extracted with EtOAc (1 L). The combined organic layers were washed with 50%> sodium bicarbonate/25%) brine (1.5 L) and dried over sodium sulfate, to give 62 g of solid. This was recrystallized from 1.8 L of 1 : 1 toluene/trimethylpentane (95 °C), to give 52.4 g of product.
[00278] 1H NMR (CDCls, 400 MHz): δ (ppm) 1.91 (s, 3H), 4.57 (dd, J= 5.12Hz and J = 12.57Hz, 1H), 4.77 (dd, J= 3.29Hz and J= 12.68Hz, 1H), 4.92-4.96 (m, 1H), 5.60 (d, J = 8.36Hz, 1H), 7.38-7.66 (m, 6H), 7.97-7.99 (m, 2H), 8.08-8.10 (m, 2H); MS (ESI) m/z= 411.1
(MNa ).
3,5-Di-0-benzoyl-2-C-chloro-2-C-methyl-D-ribofuranose (A6):
[00279] To a solution of A5 (14.48 mmol) in anhydrous tetrahydrofurane (70 ml) was added under inert atmosphere at -35°C, LiAlH(OtBu)3 (1M in tetrahydrofurane, 21.7 mmol) over a 30 min period. The reaction mixture was stirred for 1 hour at -20 °C and quenched by addition of a saturated NH4C1 solution, keeping the temperature bellow 0 °C. Ethyl acetate was added and the white suspension was filtered through a pad of celite and washed with ethyl acetate. The filtrate was extracted with ethyl acetate twice. The combined organic layers were dried over anhydrous sodium sulfate, filtered and evaporated under reduced pressure. The residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate 0 to 20%). The product was dried in vacuum (50 °C) overnight to afford expected intermediate as a colorless oil in 96% yield (mixture α/β: 45/55).
[00280] 1H NMR (CDC13, 400 MHz): δ (ppm) 1.74 (s, 1.75HP), 1.76 (s, 1.25Ha), 4.42-4.69 (m, 3H), 5.30 (d, J= 12.8Hz, 0.55HP), 5.43-5.47 (m, 0.45Ha), 5.60 (d, J= 7.0Hz, 0.55HP), 5.78 (d, J= 7.0Hz , 0.45Ha), 7.35-7.41 (m, 2H), 7.45-7.56 (m, 3H), 7.59-7.65 (m, 1H), 7.96- 8.04 (m, 2H), 8.06-8.14 (m, 2H); MS (ESI) m/z= 413 (MNa+).
3,5-Di-0-benzoyl-2-C-chloro-2-C-methyl-D-arabinofuranosyl bromide (A7):
[00281] To a solution of A6 (12.80 mmol) in anhydrous dichloromethane (80 ml) was added under inert atmosphere at -20 °C, triphenylphosphine (18.0 mmol). The reaction mixture was stirred for 15 minutes at -20 °C and CBr4 (19.20 mmol) was added. The reaction mixture was then stirred for 1 hour at -20 °C. The crude was partially concentrated under reduced pressure (bath temperature bellow 30 °C) and directly purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate 0 to 30%) to afford a mixture of β sugar A7a (1.67 g) and a sugar A7b (2.15 g) as a colorless gum in 66%> global yield.
[00282] 1H NMR (CDC13, 400 MHz): β sugar δ (ppm) 1.93 (s, 3H), 4.60-4.88 (m, 3H), 6.08 (d, J= 7.9 Hz, 1H), 6.62 (s, 1H), 7.31-7.38 (m, 2H), 7.41-7.55 (m, 3H), 7.59-7.65 (m, 1H), 8.00-8.05 (m, 2H), 8.06-8.12 (m, 2H); a sugar δ (ppm) 1.88 (s, 3H), 4.66-4.89 (m, 3H), 5.37 (d, J= 4.88Hz, 1H), 6.44 (s, 1H), 7.41-7.55 (m, 4H), 7.54-7.65 (m, 2H), 8.00-8.05 (m, 2H), 8.14-8.20 (m, 2H); MS (ESI) m/z= 476/478 (MNa+).
3 ,5′-Di-0-benzoyl-2′-C-chloro-2′-C-methyl-4-benzoyl-cytidine (A8):
[00283] To a suspension of N-benzoyl cytosine (9.48 mmol), and a catalytic amount of ammonium sulfate in 4-chlorobenzene (24 ml) was added HMDS (28.44 mmol). The reaction mixture was heated during 2 hours at 140 °C. The solvent was removed under inert atmosphere and the residue was taken in 4-chlorobenzene (15 ml). Then, A7b (4.74 mmol) in chlorobenzene (10 ml) was added dropwise to the reaction mixture followed by SnCl4 (14.22 mmol) dropwise. The reaction mixture was stirred at 70 °C overnight, cooled to room temperature and diluted with dichloromethane and a saturated NaHC03 solution. The white suspension was filtered through a pad of celite and washed with dichloromethane. The filtrate was extracted with dichloromethane twice. The combined organic layers were dried over anhydrous Na2S04, filtered and evaporated under reduced pressure to afford expected intermediate as a white solid in 89% yield.
[00284] 1H NMR (DMSO, 400 MHz): δ (ppm) 1.58 (s, 3H), 4.68-4.81 (m, 3H), 5.68 (brs, 1H), 6.55 (brs, 1H), 7.36 (d, J= 7.84 Hz, 1H), 7.39-7.76 (m, 9H), 7.88-8.07 (m, 6H), 8.30 (d, J= 7.84 Hz, 1H); MS (ESI) m/z= 588 (MH+).
3′,5′-Di-0-benzoyl-2,-C-chloro-2,-C-methyluridine (A9):
[00285] A suspension of A8 (4.19 mmol) in an acetic acid/water mixture (67 ml/17 ml, v/v), was heated at 110 °C for 3 hours. The reaction mixture was evaporated to dryness and co-evaporated with toluene (three times) to afford expected intermediate in quantitative yield as an oil which was directly used for the next step; MS (ESI) m/z= 485 (MH+). 2 -C-Chloro-2 -C-methyluridine (301):
H O CI
[00286] Intermediate A9 (4.19 mmol) in 7 N methanolic ammonia (80 ml) was stirred at room temperature for 24 hours. The mixture was evaporated to dryness, diluted with water and transferred into a separatory funnel. The aqueous layer was extracted with
dichloromethane and water was removed under reduced pressure. The residue was purified by flash RP18 gel chromatography (eluent: water/acetonitrile 0 to 40%) to afford pure expected compound as a white foam in 79% yield.
[00287] 1H NMR (DMSO, 400 MHz): δ (ppm) 1.44 (s, 3H), 3.60-3.68 (m, 1H), 3.80-3.94 (m, 3H), 5.39 (t, J= 4.45 Hz, 1H), 5.63 (d, J= 8.26 Hz, 1H), 5.93 (d, J= 5.72 Hz, 1H), 6.21 (s, 1H), 8.16 (d, J= 8.90 Hz, 1H), 11.44 (m, 1H); MS (ESI) m/z= 277 (MH+).
2′-C-Chloro-2′-C-methyl-3-benzyloxymethyluridine (Al 1):
H O CI
[00288] To a solution of 301 (0.361 mmol) in anhydrous DMF (4 ml) was added at -5 °C, DBU (0.723 mmol) followed by benzyloxymethylchloride (0.542 mmol). The reaction mixture was stirred for 45 minutes between -5 °C and 5 °C. The solvent was evaporated under reduced pressure and the residue was purified by chromatography on silica gel (eluent: dichloromethane/methanol 0 to 10%) to afford pure expected intermediate as a white solid in 80% yield.
[00289] 1H NMR (DMSO, 400 MHz): δ (ppm) 1.41 (s, 3H), 3.61-3.69 (m, 1H), 3.82-3.95 (m, 3H), 4.57 (s, 2H), 5.32 (s, 2H), 5.43 (t, J= 4.46Hz, 1H), 5.80 (d, J= 8.08Hz, 1H), 5.96 (d, J= 4.46 Hz, 1H), 6.23 (s, 1H), 7.22-7.36 (m, 5H), 8.25 (d, J= 8.22Hz, 1H); MS (ESI) m/z= 397 (MH+). Isopropyl (2S)-2-[[chloro(phenoxy)phosphoryl]amino]propanoate (A12a):
2,2-Dimethylpropyl (2S)-2-[[chloro(phenoxy)phosphoryl]amino]propanoate (A12b):
[00290] To a solution of aminoester, HC1 salt (0.434 mmol) in anhydrous dichloromethane (or acetonitrile) (4 ml) (3 times vacuo/nitrogen) under nitrogen was added at -30°C phenyldichlorophosphate (0.434 mmol) followed by N-methylimidazole (2.90 mmol)(or only 1.45 mmol for A12b). The reaction mixture was stirred at -30°C during 1 hour. The reaction was monitored by LC/MS (the sample was quenched by methanol or water) to check the complete formation of expected intermediate A12a [MS (ESI) m/z= 302 (MH+)(-OMe compounder A12b [MS (ESI) m/z= 314 (MH~)].
Compound (A13a), (A13b) or (83ii):
[00291] To the previous reaction mixture containing A12 was added All (or 302) (0.29 mmol) at -25°C under nitrogen. The reaction mixture was allowed to warm up slowly to room temperature overnight, and then diluted with dichloromethane and water (or with NaHCC”3 and EtOAc). The organic layer was extracted, dried, filtered and evaporated under reduced pressure. The crude residue was purified by chromatography on silica gel (eluent: dichloromethane/methanol 0 to 10%) (followed by preparative HPLC for A29).
Compound (A13a):
[00292] Mixture of diastereoisomers; MS (ESI) m/z= 666 (MH+). Compound (A13b):
[00293] Mixture of diastereoisomers; MS (ESI) m/z= 692.3 (MH ).
Compound (83ii):
[00294] Glassy solid; 1H NMR (CDCI3, 400MHz): δ (ppm) 1.19-1.24 (m, 9H), 1.35 (d, J = 7.1Hz, 3H), 3.95-4.05 (m, 1H), 4.31 (d, J= 8.1Hz, 2H), 4.41 (d, J= 9.0Hz, 1H), 4.59 (d, J = 7.1Hz, 2H), 4.98 (heptuplet, J= 6.28Hz, 1H), 6.38 (brs, 1H), 6.52 (s, 1H), 7.08-7.15 (m, 1H), 7.23-7.30 (m, 4H), 8.07 (s, 1H), 8.31 (s, 1H); 31P NMR (CDC13, 161.98 MHz): δ (ppm) 3.96 (s, IP); MS (ESI) m/z= 569.20 (MH+).
Compounds (40iia) and (40iib):
[00295] To a solution of A13 (0.29 mmol) in anhydrous ethanol (6 ml) was added trifluoroacetic acid (2.9 mmol) dropwise (then 3 times vacuo/nitrogen purges), followed by Palladium hydroxide (20% on Carbon). The reaction mixture was purged 3 times
vacuo/nitrogen, and 3 times vacuo/hydrogen and then stirred under hydrogen for 5 hours. The reaction mixture was diluted with ethyl acetate and filtered through a pad of celite. The filtrate was evaporated under reduced pressure, and the crude compound was purified by preparative MS/HP LC to afford two pure compounds in 48% global yield.
[00296] Compound 40ii (diastereoisomer 1): white solid; 1H NMR (CDC13, 400 MHz): δ (ppm) 1.22-1.26 (m, 6H), 1.37 (d, J= 7.08 Hz, 3H), 1.51 (s, 3H), 3.71-3.88 (m, 2H), 3.97- 4.06 (m, 1H), 4.16-4.18 (m, 1H), 4.45-4.57 (m, 2H), 4.97-5.07 (m, 1H), 5.57 (d, J= 8.20 Hz, 1H), 6.39 (s, 1H), 7.18-7.37 (m, 5H), 7.44 (d, J= 8.20 Hz, 1H), 8.40 (s, 1H); 31P NMR (CDC13, 161.98 MHz): δ (ppm) 4.20 (s, IP); MS (ESI, El+) m/z= 546 (MH+).
[00297] Compound 40ii (diastereoisomer 2): white solid; 1H NMR (CDC13, 400 MHz): δ (ppm) 1.24-1.26 (m, 6H), 1.36 (d, J= 7.04 Hz, 3H), 1.59 (s, 3H), 3.69-3.77 (m, 1H), 3.91- 3.99 (m, 2H), 4.17-4.19 (m, 1H), 4.43-4.59 (m, 2H), 5.01-5.06 (m, 1H), 5.68 (d, J= 8.20 Hz, 1H), 6.42 (s, 1H), 7.21-7.39 (m, 5H), 7.60 (d, J=8.20 Hz, 1H), 8.14 (s, 1H); 31P NMR (CDC13, 161.98 MHz): δ (ppm) 3.47 (s, IP); MS (ESI) m/z= 546 (MH+).
Compound 42ii:
[00298] Compound 42ii was synthesized from compound A13b (0.144 mmol) as described for compound 40ii.
[00299] White solid; 1H NMR (MeOD, 400 MHz) δ (ppm) 0.94 (s, 9H), 1.40 (d, J= 7.10 Hz, 3H), 1.53 (s, 3H), 3.76 (d, J= 10.43 H, 1H), 3.86 (d, J= 10.44 H, 1H), 3.98-4.06 (m, 2H), 4.18-4.22 (m, 1H), 4.39-4.44 (m, 1H), 4.52-4.57 (m, 1H), 5.62 (d, J= 8.18 Hz, 1H), 6.40 (s, 1H), 7.20-7.29 (m, 3H), 7.36-7.41 (m, 2H), 7.74 (d, J= 8.18 Hz, 1H); 31P NMR (MeOD, 161.98 MHz) δ (ppm) 3.68 (s, IP); MS (ESI) m/z = 574.08 (MH+).
PAPER
US 20170226146
https://patents.google.com/patent/US20170226146A1/en
Example 20Preparation of Compound A
- [0250]
- [0251]
A 3-neck 100 mL jacketed round bottom flask with nitrogen inlet and mechanical stirrer was charged with compound 4 (3.0 g, 10.8 mmol), compound 13 (0.484 g, 2.17 mmol, 0.20 equiv), 2-butanone (21 mL), and 2,6-lutidine (2.53 mL, 21.7 mmol, 2.0 equiv). The resulting slurry was cooled to −15° C., then a solution of compound 12 (7.96 g, 13.0 mmol) in 2-butanone (3 mL) was added over 14 hours. The reaction mixture was allowed to stir at −15° C. for an additional 25 hours and then warmed to 20° C. n-Heptane (16 mL) was added with stirring over a 1 hour period then the mixture was allowed to stir at 25° C. for 3 hours, then filtered through a fitted funnel. The filter cake was slurry-washed with a 3:2 mixture of 2-butanone and n-heptane (10 mL and then 15 mL), then dried by pulling nitrogen stream through the fritted funnel. The filter cake was slurried in a 10:1 mixture of water and 2-butanone (21 mL) and then filtered. This slurrying and filtration sequence was repeated two more times. The resulting filter cake was dried with nitrogen stream through the fritted funnel to provide compound 6.
Example 21Alternate Preparation of Compound A
- [0252]
- [0253]
Compound 6 (0.072 mmol, 1 equiv), K2HPO4 (63.0 mg, 0.361 mmol) and compound 14 (5.45 mg, 0.018 mmol) were added to a 1 dram vial with 4 A mol sieves (40 mg). To the resulting mixture was added DCM (800 μl), then the resulting reaction was allowed to stir for 5 minutes. To the reaction mixture was then added compound 14 (28.7 mg, 0.094 mmol, 1.3 equiv) and the resulting reaction was allowed to stir for about 15 hours at room temperature to provide Compound A.
Example 23Alternate Preparation of Compound A
- [0256]
- [0257]
A 100 mL reactor with nitrogen inlet and mechanical stirrer was charged with compound 4 (7.00 g, 25.3 mmol), compound 15 (0.225 g, 0.506 mmol, 0.020 equiv), 1,3-dioxolane (42 mL), and 2,6-lutidine (4.42 mL, 38.0 mmol, 1.5 equiv). The mixture was cooled to −10° C. and a 33 wt % solution of compound 12 in isopropyl acetate (29 mL, 30 mmol) was added over 1 hour. The reaction mixture was allowed to stir at −10° C. for additional 40 hours, then isopropyl acetate (28 mL) was added, and the resulting mixture was warmed to 0° C. A 10 wt % aqueous NaHSO4 solution was added (14 mL), and the mixture was allowed to stir at 30° C. for 30 minutes, then the layers were separated. To the organic layer was added an aqueous solution containing 5 wt % NaHCO3 and 5 wt % Na2SO4 (21 mL). The mixture was allowed to stir at 50° C. for 6 h. The layers were separated. To the organic layer was added 10 wt % aqueous NaCl solution (21 mL). The mixture was allowed to stir at 50° C. for 30 min. The organic layer was separated, combined with isopropyl acetate (5 mL) and concentrated in vacuo to half volume at 20000 pa in a 50° C. bath. The resulting solution was solvent-switched with isopropanol (4×35 mL) to 60 g weight. The mixture was seeded with 100 mg of compound A at 60° C. The resulting slurry was allowed to stir at 55° C. for 30 minutes, then n-Heptane (35 mL) was added over 1 hour at 55° C. The resulting slurry was allowed to stir for an additional 1 hour at 55° C., then cooled to room temperature and filtered. The filter cake was washed with a 1:1 mixture of isopropanol and n-heptane (3×14 mL), followed by n-heptane (14 mL), then dried under nitrogen to provide Compound A.
PAPER
https://pubs.rsc.org/en/content/articlelanding/2021/sc/d1sc01978c#!divAbstract
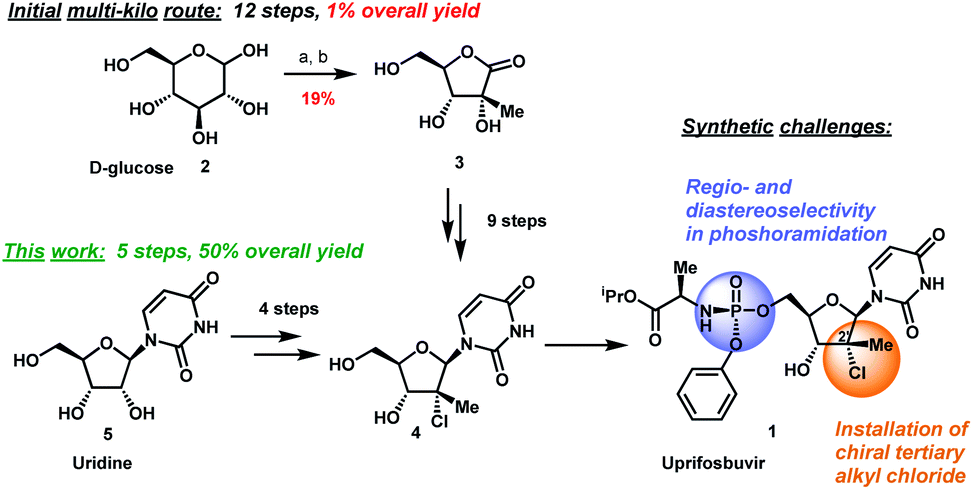
Uprifosbuvir is an antiviral agent developed for treatment of chronic hepatitis C infections. Its original synthesis route requires twelve steps with an overall yield of only 1 %. Such a difficult and time-consuming synthesis approach is acceptable for the early trial phase of a new drug, but impractical for broad application as hepatitis C treatment or for repurposing against novel viral diseases.
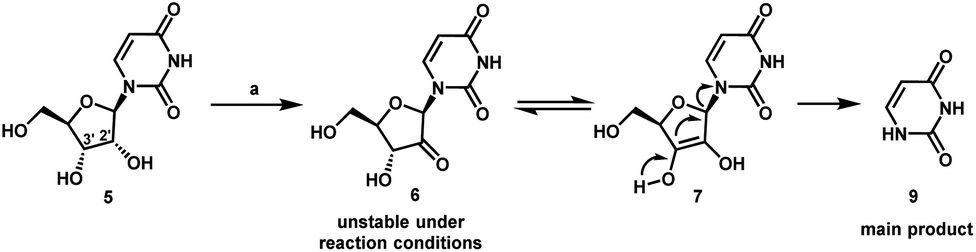
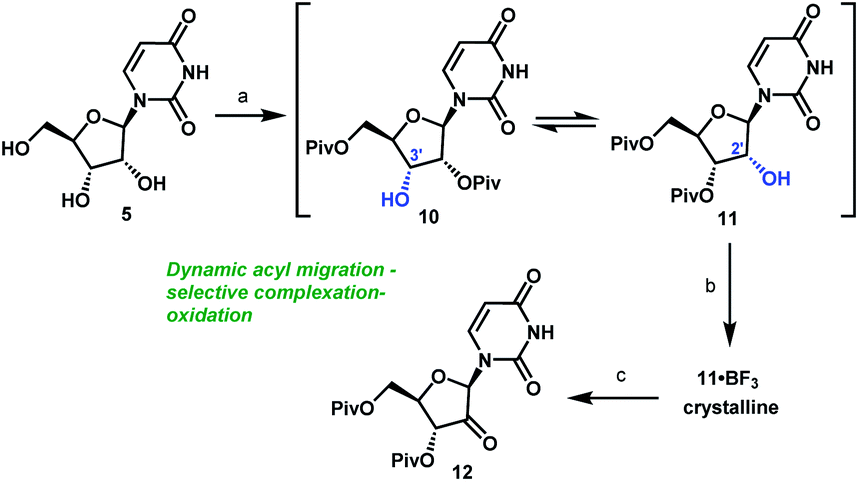
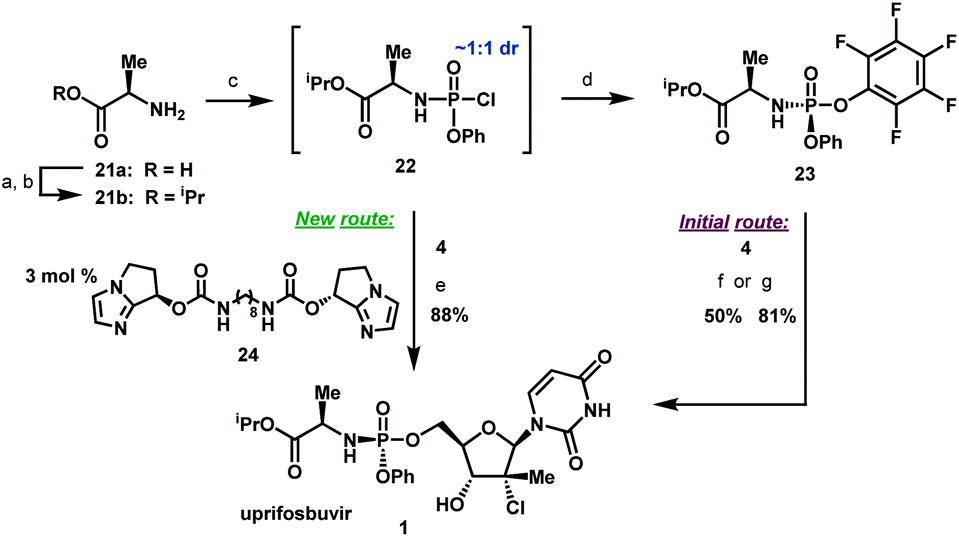
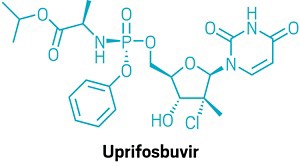
Artis Klapars, John Y. L. Chung, and colleagues, Merck & Co., Inc., Rahway, NJ, USA, and WuXi STA, Shanghai, China, have developed a synthesis route for uprifosbuvir requiring only five steps and starting from readily available uridine. Initially, uridine is selectively oxidized after OH-acylation with pivaloyl chloride in an acyl migration/oxidation process driven by complexation with the Lewis acid BF3*OEt2 in toluene. In the second step, methylation is achieved by MeMgBr/MgCl2 in a toluene/anisole mixture where a more reactive methyl-manganese species is formed in-situ from the Grignard reagent, providing high yield and a good diastereomeric ratio (dr). Subsequently, the tertiary chloride group is introduced. Due to the high functional-group density, a cyclodehydration step is required before chlorination to avoid side reactions. The chlorination is carried out using dichlorodimethylsilane with FeCl3*6H2O and tetramethyldisiloxane as additives which avoids the hazardous use of HCl gas under pressure required in the initial synthesis. In the final step, the regioselective phosphoramidation is achieved using a chlorophosphoramidate precursor and a dimeric chiral imidazole carbamate catalyst which led to a dr of 97:3 starting from a 1:1 diastereomeric mixture of the chlorophosphoramidate reagent.
Uprifosbuvir was synthesized with an overall yield of 50 %, a vast improvement compared to the 1 % of the original synthesis route. Additionally, the newly developed synthesis steps have the potential to provide easier access to other nucleoside-based antiviral agents.
- Efficient Synthesis of Antiviral Agent Uprifosbuvir Enabled by New Synthetic Methods,
Artis Klapars, John Chung, John Limanto, Ralph Calabria, Louis-Charles Campeau, Kevin Campos, Wenyong Chen, Stephen M Dalby, Tyler A Davis, Daniel DiRocco, Alan Hyde, Amude M Kassim, Mona Utne Larsen, Guiquan Liu, Peter Maligres, Aaron Moment, Feng Peng, Rebecca Ruck, Michael Shevlin, Bryon L Simmons, Zhiguo Jake Song, Lushi Tan, Timothy J Wright, Susan Zultanski,
Chemical Science 2021.
https://doi.org/10.1039/D1SC01978C
Efficient synthesis of antiviral agent uprifosbuvir enabled by new synthetic methods†
This article is Open Access
All publication charges for this article have been paid for by the Royal Society of Chemistry
Abstract
An efficient route to the HCV antiviral agent uprifosbuvir was developed in 5 steps from readily available uridine in 50% overall yield. This concise synthesis was achieved by development of several synthetic methods: (1) complexation-driven selective acyl migration/oxidation; (2) BSA-mediated cyclization to anhydrouridine; (3) hydrochlorination using FeCl3/TMDSO; (4) dynamic stereoselective phosphoramidation using a chiral nucleophilic catalyst. The new route improves the yield of uprifosbuvir 50-fold over the previous manufacturing process and expands the tool set available for synthesis of antiviral nucleotides.
Scheme 1 Synthetic approaches to uprifosbuvir
1 with the two main challenges highlighted. (a) Me
2NH, AcOH, EtOH/MeOH, 80 °C, 1.5 h; (b) Ca(OH)
2, water, 70 °C, 24 h, 19% over 2 steps.
9
Scheme 3 Complexation-driven selective acyl migration/oxidation to access 12. (a) PivCl, pyridine, 0 °C, 16 h; (b) BF3·OEt2, PhMe, 40 °C, 10 h; (c) TEMPO, Bu4NBr, AcOOH, dioctyl sulphide, PhMe, −10 °C to 20 °C, 24 h, 83% from 5.
Scheme 6 Completion of uprifosbuvir synthesis. (a) TMS-Cl, iPrOH, 70 °C, 12 h; (b) NEt
3, iPrOAc, wiped film evaporation, 80%; (c) PhOP(O)Cl
2, NEt
3, iPrOAc, −20 °C, 2 h, 90%; (d) C
6F
5OH, NEt
3, iPrOAc, −5 °C to 10 °C, 18 h, 76%;
26 (e)
4, 3 mol%
24, 2,6-lutidine, 1,3-dioxolane, −10 °C, 24 h, 88%; (f)
4, tBuMgCl, THF, −5 °C to 5 °C, 15 h, 50%;
27 (g)
4, Me
2AlCl, 2,6-lutidine, THF, 35 °C, 16 h, 81%.
27
Scheme 7 Summary of uprifosbuvir synthesis. AY = assay yield; IY = isolated yield.
PAPER
References
- ^ Soriano V, Fernandez-Montero JV, de Mendoza C, Benitez-Gutierrez L, Peña JM, Arias A, Barreiro P (August 2017). “Treatment of hepatitis C with new fixed dose combinations”. Expert Opinion on Pharmacotherapy. 18 (12): 1235–1242. doi:10.1080/14656566.2017.1346609. PMID 28644739. S2CID 205819421.
- ^ Borgia G, Maraolo AE, Nappa S, Gentile I, Buonomo AR (March 2018). “NS5B polymerase inhibitors in phase II clinical trials for HCV infection”. Expert Opinion on Investigational Drugs. 27 (3): 243–250. doi:10.1080/13543784.2018.1420780. PMID 29271672. S2CID 3672885.
- ^ Lawitz E, Gane E, Feld JJ, Buti M, Foster GR, Rabinovitz M, et al. (September 2019). “Efficacy and safety of a two-drug direct-acting antiviral agent regimen ruzasvir 180 mg and uprifosbuvir 450 mg for 12 weeks in adults with chronic hepatitis C virus genotype 1, 2, 3, 4, 5 or 6”. Journal of Viral Hepatitis. 26 (9): 1127–1138. doi:10.1111/jvh.13132. PMID 31108015. S2CID 160014275.
Uprifosbuvir (MK-3682) is an antiviral drug developed for the treatment of Hepatitis C. It is a nucleotide analogue which acts as an NS5B RNA polymerase inhibitor. It is currently in Phase III human clinical trials.[1][2][3]
References
- ^ Soriano V, Fernandez-Montero JV, de Mendoza C, Benitez-Gutierrez L, Peña JM, Arias A, Barreiro P (August 2017). “Treatment of hepatitis C with new fixed dose combinations”. Expert Opinion on Pharmacotherapy. 18 (12): 1235–1242. doi:10.1080/14656566.2017.1346609. PMID 28644739. S2CID 205819421.
- ^ Borgia G, Maraolo AE, Nappa S, Gentile I, Buonomo AR (March 2018). “NS5B polymerase inhibitors in phase II clinical trials for HCV infection”. Expert Opinion on Investigational Drugs. 27 (3): 243–250. doi:10.1080/13543784.2018.1420780. PMID 29271672. S2CID 3672885.
- ^ Lawitz E, Gane E, Feld JJ, Buti M, Foster GR, Rabinovitz M, et al. (September 2019). “Efficacy and safety of a two-drug direct-acting antiviral agent regimen ruzasvir 180 mg and uprifosbuvir 450 mg for 12 weeks in adults with chronic hepatitis C virus genotype 1, 2, 3, 4, 5 or 6”. Journal of Viral Hepatitis. 26 (9): 1127–1138. doi:10.1111/jvh.13132. PMID 31108015. S2CID 160014275.
//////////uprifosbuvir, MK 3682, ウプリホスブビル,
уприфосбувир, أوبريفوسبوفير ,
乌磷布韦 , IDX-21437,
DB15206,
SB18784,
D10996,
Q27281714, IDX 21437, PHASE 3
CC(C)OC(=O)C(C)NP(=O)(OCC1C(C(C(O1)N2C=CC(=O)NC2=O)(C)Cl)O)OC3=CC=CC=C3




































 *a
*a