














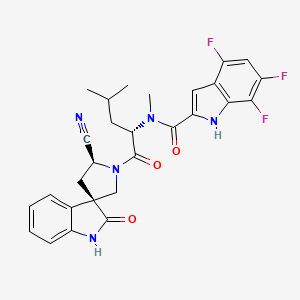
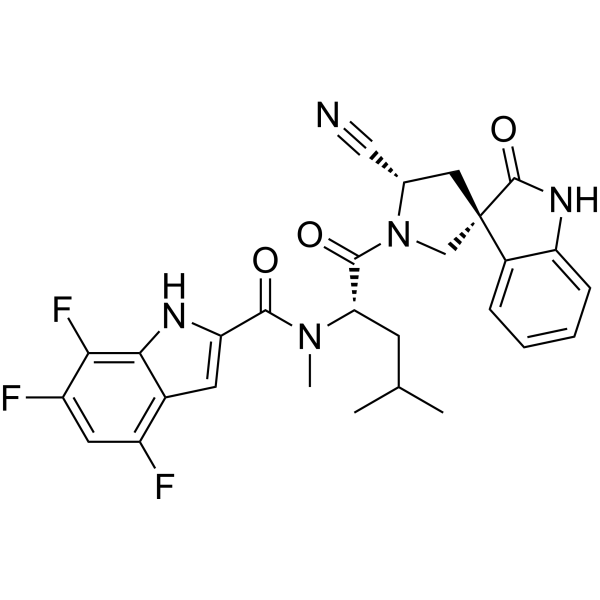
Zevotrelvir, EDP 235
cas 2773516-53-1
N-[(2S)-1-[(2′S,3R)-2′-cyano-2-oxospiro[1H-indole-3,4′-pyrrolidine]-1′-yl]-4-methyl-1-oxopentan-2-yl]-4,6,7-trifluoro-N-methyl-1H-indole-2-carboxamide
C28H26F3N5O3, 537.5
Zevotrelvir (Compound 52) is a coronavirus inhibitor with IC50 ranges of <0.1 μM and <0.1mM for 229E hCoV and SARS-CoV-23C-like (3CL) proteases, respectively. Zevotrelvir has the potential to study viral infections.
Coronaviruses are enveloped, positive-sense, single-stranded RNA viruses. The genomic RNA of CoVs has a 5′-cap structure and 3′-poly-A tail and contains at least 6 open reading frames (ORFs). The first ORF (ORF 1a/b) directly translates two polyproteins: pp1a and pp1ab. These polyproteins are processed by a 3C-Like protease (3CLpro), also known as the main protease (Mpro), into 16 non-structural proteins. These non-structural proteins engage in the production of subgenomic RNAs that encode four structural proteins, namely envelope, membrane, spike, and nucleocapsid proteins, among other accessory proteins. As a result, it is understood that 3C-Like protease has a critical role in the coronavirus life cycle.
3CLpro is a cysteine protease involved in most cleavage events within the precursor polyprotein. Active 3CLpro is a homodimer containing two protomers and features a Cys-His dyad located in between domains I and II.3CLpro is conserved among coronaviruses and several common features are shared among the substrates of 3CLpro in different coronaviruses. As there is no human homolog of 3CLpro, it is an ideal antiviral target. Although compounds have been reported to inhibit 3CLpro activity, they have not been approved as coronavirus therapies. (Refer to
WO 2004101742 A2, US 2005/0143320 Al, US 2006/0014821 Al, US 2009/0137818
Al, WO 2013049382 A2, WO 2013166319 A1, WO2018042343, WO2018023054, WO 2022013684, WO 2021252644, WO2022020711, WO 2022020242, US 11,174,231 B1, US 11,124,497 B1, WO 2005113580, and WO2006061714).
There is a need in the art for novel therapeutic agents that treat, ameliorate or prevent SARS-CoV-2 infection. The present invention provides the process of novel compounds which act in inhibiting or preventing SARS-CoV-2 viral replication and thus are used in the treatment of COVID-19 (see PCT/US21/60247).
Synthesis of substituted spirooxindole and its intermediate has been previously published (Refer to PCT/US21/60247, WO2019086142, WO 2020221811, WO2020221826, J. Med. Chem.2012, 55, 9069). However, the scale-up using previous process is very challenging due to the safety concern associated with certain intermediates, instability of certain intermediates as well as lack of purification process other than column chromatograph. Thus, there is a strong need for developing a safe and efficient processes for the large-scale preparation of these novel substituted spirooxindole derivatives.
SYNTHESIS

https://patents.google.com/patent/US11352363B1/en

PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US362064348&_cid=P10-M0SZPD-12620-1
SYN
[1]. Guoqiang Wang, et al. Novel spiropyrrolidine derived antiviral drugs. Patent CN114524821A.
1.20230295175PROCESSES FOR THE PREPARATION OF SUBSTITUTED SPIROOXINDOLE DERIVATIVES
2.WO/2023/177854PROCESSES FOR THE PREPARATION OF SUBSTITUTED SPIROOXINDOLE DERIVATIVES
3.WO/2022/109363NOVEL SPIROPYRROLIDINE DERIVED ANTIVIRAL AGENTS
Enanta Pharmaceuticals, Inc.
WO2023177854
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023177854&_cid=P10-M0SZ7T-99709-1




Example 15. Preparation of Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))

DMF (760 kg, 8V) was added into the reaction at 0 °C (-5~5 °C) followed by compound (j) (63 kg, 1.05 eq) and N-Methylmorpholine (56 kg, 2 eq), HATU
(106 kg, 1.0 eq) and Compound (m-1) (100 kg, 1.0 eq). The reactor was rinsed with DMF (190 kg, 2V) under and warmed up to 25 °C (20~30 °C) and stirred for 5 h (3~6 h) at 25 °C (20~30 °C). After that, additional HATU (0.1 eq) was added and the reaction mixture was stirred for 16-24 h.25% Ammonium hydroxide (38 kg) was added to the reaction mixture at 25 °C (20~30 °C) and stirred for 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was then added to water (5000 kg, 50V) at 20-30°C over 2 h and the resulting slurry was stirred for 2 h (1~5 h) at 25 °C (20~30 °C). The mixture was filtered and the cake was rinsed with water (500 kg, 5 V). The cake was dissolved in ethyl acetate (1350 kg, 15 V) and washed with 10% sodium chloride solution (500 kg) for three times. The organic layer was separated to 1.5-2.5V at not more than 45℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (660 kg, 5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (137 kg, 2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and the wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in 80-85% yield.
1H NMR (300 MHz, DMSO-d6) δ 12.46 (s, 1H), 10.68 (s, 1H), 7.56 (s, 1H), 7.15 – 7.00 (m, 3H), 6.91 (t, J = 4.4 Hz, 2H), 6.84 (d, J = 7.7 Hz, 1H), 6.55 (d, J = 2.8 Hz, 1H), 5.34 (t, J = 7.3 Hz, 1H), 4.63 (dd, J = 9.8, 8.0 Hz, 1H), 3.83 (q, J = 10.3 Hz, 2H), 3.45 (qd, J = 7.0, 5.1 Hz, 1H), 3.16 (s, 3H), 2.35 – 2.13 (m, 2H), 1.69 (t, J = 7.1 Hz, 2H), 1.56 (dq, J = 13.1, 6.5 Hz, 1H), 0.93 (dd, J = 12.2, 6.3 Hz, 6H).
Example 16. Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))
DMF solution of Compound (m-2) (1 kg, 1.0 eq.) was added to a reactor at around 0-10oC. Compound (l) (600 g, 1.0 eq.), NMM (3.00 eq., 850 g) and HATU (1.00 eq., 1.06 kg) was added to the reactor while maintaining the temperature at 0-10oC; The reaction was warmed to 20±5oC, and stirred for at least 6 hours at 20±5oC. HATU (0.20 eq., 210 g) was added to the reactor at 20±5oC and stirred for at least 6 hours at 20±5oC.25% Ammonium hydroxide (390 g, 1.0 eq) was added to the reaction mixture at 20 °C and stirred for 2 h (1~3 h) at 20 °C. EtOAc (14.0 V) and water (14 V) was added at around 25oC over 20 minutes, and the
solution was stirred for at least 30 min. Aqueous phase was extracted with EtOAc for three times and the organic phase was combined, and washed with 10% aq. NaCl for three times at 20±5oC. The organic phase was concentrated to 6 V then EtOH (7.0 V) was charged. The EtOAc-EtOH solvent swap was repeated for three times and concentrated to 5 V before water (7.0 v) was added at 20±5oC. The mixture was cooled to 0-10oC and stirred for 1 h before being filtered. The filter cake was dissolved in ethyl acetate (15 V) and washed with 10% sodium chloride solution for three times. The organic layer was concentrated to 2-3V at not more than 45℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in about 70-75% yield over two steps.
1H NMR (300 MHz, DMSO-d6) δ 12.46 (s, 1H), 10.68 (s, 1H), 7.56 (s, 1H), 7.15 – 7.00 (m, 3H), 6.91 (t, J = 4.4 Hz, 2H), 6.84 (d, J = 7.7 Hz, 1H), 6.55 (d, J = 2.8 Hz, 1H), 5.34 (t, J = 7.3 Hz, 1H), 4.63 (dd, J = 9.8, 8.0 Hz, 1H), 3.83 (q, J = 10.3 Hz, 2H), 3.45 (qd, J = 7.0, 5.1 Hz, 1H), 3.16 (s, 3H), 2.35 – 2.13 (m, 2H), 1.69 (t, J = 7.1 Hz, 2H), 1.56 (dq, J = 13.1, 6.5 Hz, 1H), 0.93 (dd, J = 12.2, 6.3 Hz, 6H).
Example 17. Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))
DMF (10.0 v) was added to a reactor at 25 °C followed by Compound (l) (4.4 kg, 1.0 eq.), NMM (3.0 eq.) Compound (m-3) (1.0 eq.) and HATU (1.0 eq) at 20-25oC. The reaction mixture was stirred for at least 12 hours at 20-25 °C. Once reaction was complete, aqueous ammonium hydroxide (1.0 eq.) was to the reaction system at 20-25 °C, then stirred for at least 2 hours at 20-25oC. The reaction mixture was then added to water (220 kg, 50V) at 20-30°C over 2 h and the resulting slurry was stirred for 2 h (1~5 h) at 25 °C (20~30 °C). The mixture was filtered and the cake was rinsed with water (22 kg, 5 V). The cake was dissolved in ethyl acetate (135 g, 15 V) and washed with 10% sodium chloride solution (22 kg) for three times. The organic layer was separated to 1.5-2.5V at not more than 45 ℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in 80-85% yield.
1H NMR (300 MHz, DMSO-d6) δ 12.46 (s, 1H), 10.68 (s, 1H), 7.56 (s, 1H), 7.15 – 7.00 (m, 3H), 6.91 (t, J = 4.4 Hz, 2H), 6.84 (d, J = 7.7 Hz, 1H), 6.55 (d, J = 2.8 Hz, 1H), 5.34 (t, J = 7.3 Hz, 1H), 4.63 (dd, J = 9.8, 8.0 Hz, 1H), 3.83 (q, J = 10.3 Hz, 2H), 3.45 (qd, J = 7.0, 5.1 Hz, 1H), 3.16 (s, 3H), 2.35 – 2.13 (m, 2H), 1.69 (t, J = 7.1 Hz, 2H), 1.56 (dq, J = 13.1, 6.5 Hz, 1H), 0.93 (dd, J = 12.2, 6.3 Hz, 6H). Example 18. Preparation of N-((S)-1-((3R,5’S)-5′-cyano-2-oxospiro[indoline-3,3′-pyrrolidin]-1′-yl)-4-methyl-1-oxopentan-2-yl)-4,6,7-trifluoro-N-methyl-1H-indole-2-carboxamide toluene solvate (Compound (I))
(I))

Ethyl acetate (630 kg, 10 V) was added into reactor (R1) followed by Compound (n) (70 kg). Make sure the water content was less than 0.20% (w/w). The reaction was cooled to 0 °C (-5 – 5°C) and then triethylamine (89.6 kg) was added followed by trifluoroacetic anhydride (92.4 kg) at 0 °C (-5 – 5°C). The reaction was stirred for 1 h (0.5~2 h) at 0 °C (-5 – 5°C). Once the reaction was complete, the reaction mixture was added slowly to 0.2 N aqueous HCl solution (700 kg) over 1 h at 0 °C (-5~5 °C). The resulting solution was stirred for 30 min at 0 °C (-5~5 °C) and the organic layer was separated.1% aqueous ammonium hydroxide (700 kg) was added to the organic layer and stirred at 20 °C for 30 min (15~25 °C). The organic layer was separated and washed with 10% brine for four times. Then the organic layer was separated and distilled to 2-3 V. Toluene-EtOAc swap was performed until precipitate was observed at 3-4 V. Then Toluene (5-6 V) was added and the slurry was stirred at 50 oC for 2 h. Then the solution was cooled down to 20 oC over 1-2 h and stirred for 10 hr (6~14 hr) at 20 °C (15~25 °C). The reaction mixture was filtered and the wet cake was rinsed with toluene (120 kg, 2V). The wet cake was then dried at 50˚C (45~55 °C) for 48 hr to provide desired compound (o) as a white solid in 80-85% yield.
1H NMR (400 MHz, Acetone-d6) δ 11.17 (s, 1H), 9.65 (s, 1H), 7.02 (dd, J = 13.7, 7.3 Hz, 2H), 6.94 (dd, J = 6.0, 3.5 Hz, 1H), 6.92 – 6.85 (m, 2H), 6.81 (t, J = 7.5 Hz, 1H), 5.56 (dd, J = 9.4, 5.6 Hz, 1H), 5.21 (t, J = 8.3 Hz, 1H), 4.25 (d, J = 10.7 Hz, 1H), 3.99 (d, J = 10.6 Hz, 1H), 3.43 (s, 3H), 2.79 – 2.61 (m, 2H), 1.93 (ddd, J = 14.4, 9.5, 5.1 Hz, 1H), 1.79 (ddd, J = 14.2, 8.7, 5.6 Hz, 1H), 1.64 (dpd, J = 8.7, 6.6, 5.1 Hz, 1H), 0.98 (dd, J = 18.5, 6.6 Hz, 6H).
US20230103494
CN114524821
SCHEME

MAIN

////////Zevotrelvir, EDP 235
O=C1[C@@]2(CN([C@@H](C2)C#N)C([C@H](CC(C)C)N(C)C(C3=CC4=C(F)C=C(F)C(F)=C4N3)=O)=O)C5=CC=CC=C5N1