














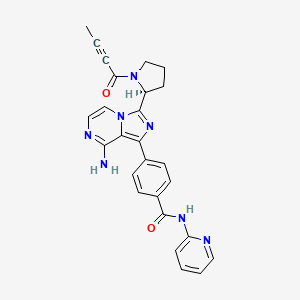
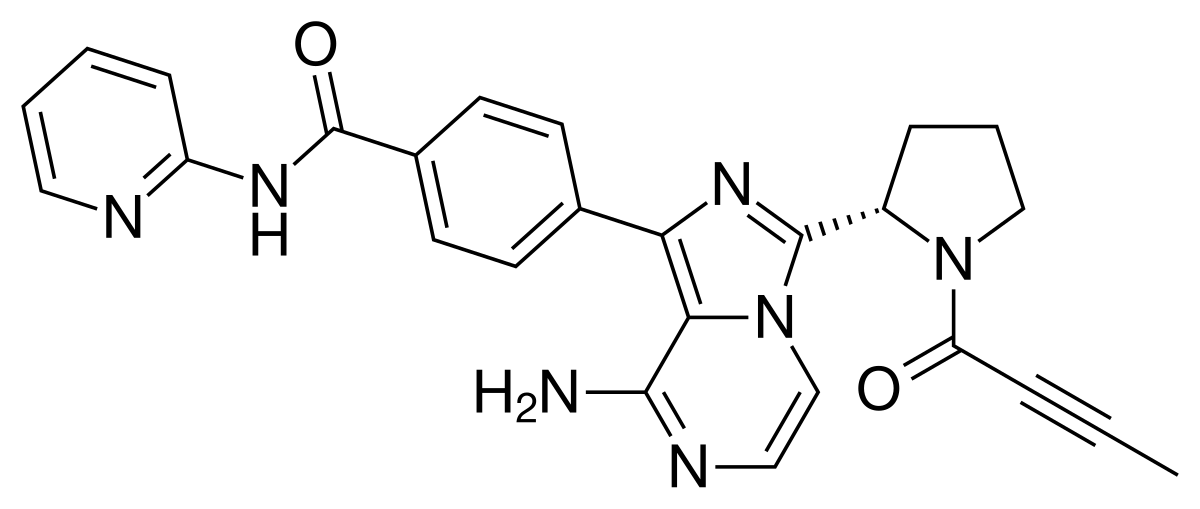
Acalabrutinib
- Molecular FormulaC26H23N7O2
- Average mass465.507 Da
Acalabrutinib, rINN, ACP-196,
FDA 2017 APPROVED, Lymphoma, mantle cell, ACERTA PHARMA
Orphan Drug, breakthrough therapy designation,
(S)-4-[8-Amino-3-[1-(but-2-ynoyl)pyrrolidin-2-yl]imidazo[1,5-a]pyrazin-1-yl]-N-(pyridin-2-yl)benzamide
(S)-4-(8-amino-3-n-but-2-vnoylpyrrolidin-2-vnimidazo[1 ,5-alpyrazin-1-yl)-N-(pyridin-2-yl)benzamide

Acalabrutinib (rINN,[1] ACP-196) is a novel experimental anti-cancer drug and a 2nd generation Bruton’s tyrosine kinase (BTK) inhibitor[2][3] developed by Acerta Pharma.[4] It is more potent and selective (fewer side-effects) than ibrutinib, the first-in-class BTK inhibitor.[2][3][5]
The compound was granted orphan drug designation for the treatment of chronic lymphocytic leukemia, Waldenström’s macroglobulinemia and mantle cell lymphoma in the U.S. and the E.U. in 2015 and 2016, respectively. In 2017, the product was granted breakthrough therapy designation in the U.S. for the treatment of patients with mantle cell lymphoma who have received at least one prior therapy.
Acalabrutinib is an orally available inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Upon administration, acalabrutinib inhibits the activity of BTK and prevents the activation of the B-cell antigen receptor (BCR) signaling pathway. This prevents both B-cell activation and BTK-mediated activation of downstream survival pathways. This leads to an inhibition of the growth of malignant B cells that overexpress BTK. BTK, a member of the src-related BTK/Tec family of cytoplasmic tyrosinekinases, is overexpressed in B-cell malignancies; it plays an important role in B lymphocyte development, activation, signaling, proliferation and survival.
Acalabrutinib is a Bruton’s Tyrosine Kinase (BTK) inhibitor developed at Acerta Pharma launched in 2017 in the U.S. for the oral treatment of adults with mantle cell lymphoma who have received at least one prior therapy.
-Facebook.png)


Clinical and Regulatory Status
Pre-clinical
Relative to ibrutinib, acalabrutinib demonstrated higher selectivity and inhibition of the targeted activity of BTK, while having a much greater IC50 or otherwise virtually no inhibition on the kinase activities of ITK, EGFR, ERBB2, ERBB4, JAK3, BLK, FGR, FYN, HCK, LCK, LYN, SRC, and YES1.[3] In addition, in platelets treated with ibrutinib, thrombus formation was clearly inhibited while no impact to thrombus formation was identified relative to controls for those treated with acalabrutinib.[3] These findings strongly suggest an improved safety profile of acalabrutinib with minimized adverse effects relative to ibrutinib.[3]
As was conducted in the development of ibrutinib, pre-clinical studies of acalabrutinib included in vitro and in vivo pharmacodynamic evaluation in a canine lymphoma model.[6] A dose-dependent relationship resulting in cyto-toxicity and anti-proliferative effects was first demonstrated in a canine lymphoma cell line in vitro.[6] In vivo, the compound was found to be generally safe and well tolerated in the dosage range of 2.5–20 mg/kg every 12 or 24 hours, with clinical benefit observed in 30% of canine patients while observed adverse events consisted primarily of gastrointestinal effects such as anorexia, weight loss, vomiting, diarrhea and lethargy.[6]

Clinical
The interim results of the still on-going first human phase 1/2 clinical trial (NCT02029443) with 61 patients for the treatment of relapsed chronic lymphocytic leukemia (CLL) are encouraging, with a 95% overall response rate demonstrating potential to become a best-in-class treatment for CLL.[2][7] Notably, a 100% response rate was achieved for those patients which were positive for the 17p13.1 gene deletion – a subgroup of patients that typically results in a poor response to therapy and expected outcomes.[3]
The most common adverse events were headache, diarrhea and weight gain.[3] Despite the appearance of a greater occurrence of transient headaches, the pre-clinical data suggests a preferred advantage of acalabrutinib over ibrutinib due to expected reduced adverse events of skin rash, severe diarrhea, and bleeding risk.[3] An additional clinical trial is currently in progress to directly compare the safety and efficacy performance of acalabrutinib to ibrutinib to better elucidate the differences in the therapeutic agents.[3]
While the primary indication is for CLL, as of late 2016, acalabrutinib is under evaluation for multiple indications in 20+ clinical trials (alone and in combination with other interventions) for various blood cancers, solid tumors, and rheumatoid arthritis.[7][8] Approximately 1,000 patients have been treated with acalabrutinib in clinical trials so far, including more than 600 on acalabrutinib alone and almost 400 on additional therapies in combination with acalabrutinib.[9]
Regulatory
As of February 2016, acalabrutinib had received orphan designation in the United States for CLL only,[10] and was similarly designated as an orphan medicinal product by the European Medicines Agency (EMA) Committee for Orphan Medicinal Products (COMP) for treatment of three indications – chronic lymphocytic leukemia (CLL)/ small lymphocytic lymphoma (SLL), mantle cell lymphoma (MCL), and lymphoplasmacytic lymphoma (Waldenström’s macroglobulinaemia, MG).[11] If the drug is ultimately approved, this designation will result in a 10-year period of market exclusivity for the stated indications within Europe.[12]
Commercial Aspects
Acerta Pharma, the innovator responsible for the discovery and development of acalabrutinib, is a clinical stage biopharmaceutical company recently founded in 2012 in Oss, the Netherlands.[13] A combined $13 Million in Series A funding was secured March 14, 2013 from various investor sources including the venture capital firms of BioGeneration Ventures and OrbiMed Advisors, the Dutch State and Province of Brabant through the Brabant Development Agency, and the private US equity firm Frazier Healthcare.[14] Further undisclosed amounts of Series B funding was secured May 2015 from the mutual fund company T. Rowe Price.[15]
After the promising results for the treatment of CLL in initial clinical trials,[2] Astra Zeneca purchased a 55% stake in Acerta Pharma for $4 billion in December 2015, with an option to acquire the remaining 45% stake for an additional $3 billion, conditional on the first approval in both the US and Europe and the establishment of commercial opportunity.[16]
Intellectual Property
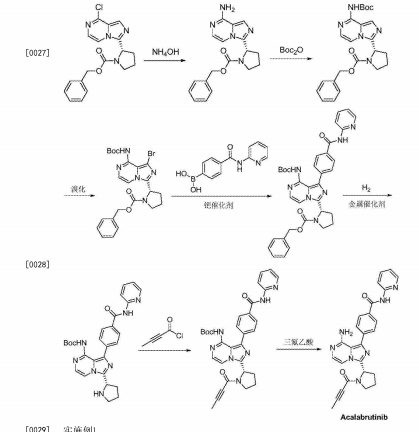
Several patents have been filed by Acerta Pharma through the World Intellectual Property Organization (WIPO) for the use of acalabrutinib (and structurally similar derivatives) either alone or in combination with additional therapeutic agents for the treatment of various hematological and solid tumor cancers as well as inflammatory and autoimmune diseases.[17][18][19][19][20][21][22][23][24][25][26][27]
Notably, patents filed through WIPO still need to be filed appropriately for each individual nation on the path to commercialization. For example, one related United States patent application is US2014155385, which was filed July 11, 2012 and approved June 5th, 2014 for the use of 6-5 membered fused pyridine ring compounds (including acalabrutnib and its structurally similar derivatives) in the treatment of BTK mediated disorders.[28]
SYNTHESIS
WO 2013010868

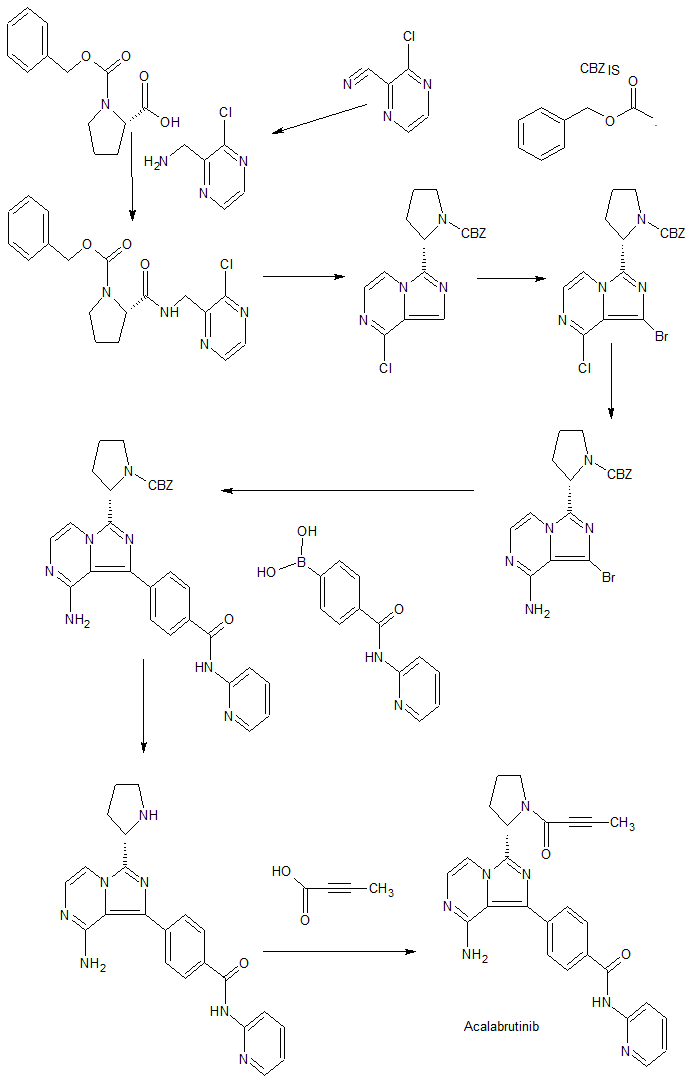
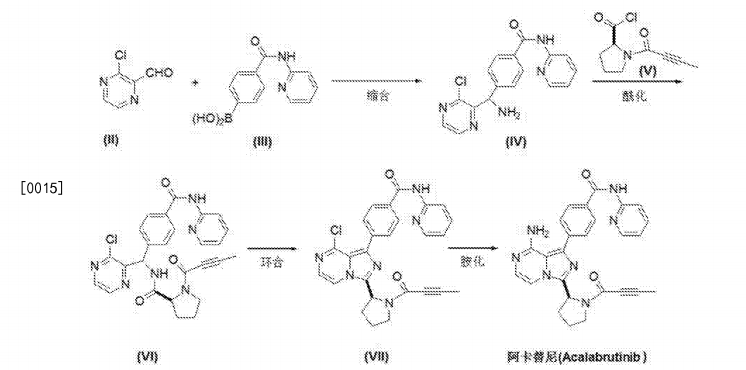
Synthesis of acalabrutinib, using 3-chloropyrazine-2-carbonitrile as the starting material, is described. The method comprises reduction of the starting material, condensation with N-Cbz-L-proline, intramolecular cyclization, bromination, Suzuki coupling with (4-(2-pyridylcarbamoyl)phenyl)boronic acid and condensation with 2-butynoic acid. WO 2013010868
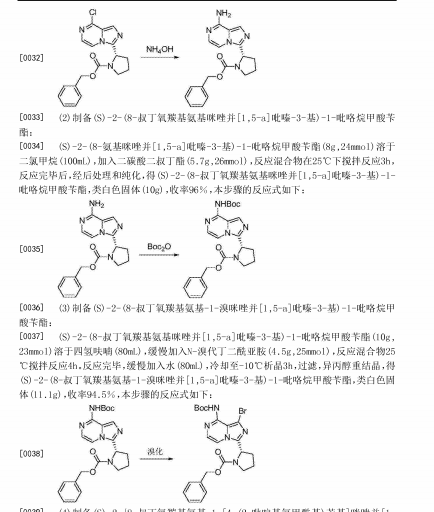
Reduction of 3-chloropyrazine-2-carbonitrile with H2 over Raney-Ni in AcOH, followed by treatment with aqueous HCl in Et2O gives (3-chloro-2-pyrazinyl)methylamine hydrochloride , which upon condensation with N-Cbz-L-proline in the presence of HATU and Et3N in CH2Cl2 affords amide .
Intramolecular cyclization of intermediate by means of DMI and POCl3 in acetonitrile at 63 °C provides N-Cbz-8-chloro-3-[2(S)-pyrrolidinyl]imidazo[1,5-a]pyrazine , which is brominated with NBS in DMF to yield N-Cbz-1-bromo-8-chloro-3-[2(S)-pyrrolidinyl]imidazo[1,5-a]pyrazine .
Reaction of chloro compound with NH3 in i-PrOH at 110 °C produces N-Cbz-1-bromo-3-[2(S)-pyrrolidinyl]imidazo[1,5-a]pyrazin-8-amine , which upon Suzuki coupling with (4-(2-pyridylcarbamoyl)phenyl)boronic acid in the presence of PdCl2(dppf) and K2CO3 in dioxane at 140 °C under microwave irradiation furnishes diaryl derivative .
Removal of the benzyloxycarbonyl moiety in intermediate using HBr in AcOH generates pyrrolidine derivative , which is condensed with 2-butynoic acid in the presence of HATU and Et3N in CH2Cl2 to afford the target acalabrutinib
PATENT
WO 2013010868
https://www.google.com/patents/WO2013010868A1?cl=en
scheme I

scheme II

Intermediate 1

(S)-Benzyl 2-(8-amino-1-bromoimidazo[1 ,5-alpyrazin-3-vnpyrrolidine-1-carboxylate
(a) (3-Chloropyrazin-2-yl)methanamine. hydrochloride
To a solution of 3-chloropyrazine-2-carbonitrile (160 g, 1 .147 mol) in acetic acid (1.5 L) was added Raney Nickel (50% slurry in water, 70 g, 409 mmol). The resulting mixture was stirred under 4 bar hydrogen at room temperature overnight. Raney Nickel was removed by filtration over decalite and the filtrate was concentrated under reduced pressure and co-evaporated with toluene. The remaining brown solid was dissolved in ethyl acetate at 50°C and cooled on an ice-bath. 2M hydrogen chloride solution in diethyl ether (1 .14 L) was added in 30 min. The mixture was allowed to stir at room temperature over weekend. The crystals were collected by filtration, washed with diethyl ether and dried under reduced pressure at 40°C. The product brown solid obtained was dissolved in methanol at 60°C. The mixture was filtered and partially concentrated, cooled to room temperature and diethyl ether (1000 ml) was added. The mixture was allowed to stir at room temperature overnight. The solids formed were collected by filtration, washed with diethyl ether and dried under reduced pressure at 40°C to give 153.5 g of (3-chloropyrazin-2- yl)methanamine. hydrochloride as a brown solid (74.4 %, content 77 %).
(b) (S)-benzyl 2-((3-chloropyrazin-2-yl)methylcarbamoyl)pyrrolidine-1-carboxylate
To a solution of (3-chloropyrazin-2-yl)methanamine.HCI (9.57 g, 21.26 mmol, 40% wt) and Z-Pro-OH (5.3 g, 21 .26 mmol) in dichloromethane (250 mL) was added triethylamine (1 1.85 mL, 85 mmol) and the reaction mixture was cooled to 0°C. After 15 min stirring at 0°C, HATU (8.49 g, 22.33 mmol) was added. The mixture was stirred for 1 hour at 0°C and then overnight at room temperature. The mixture was washed with 0.1 M HCI-solution, 5% NaHC03, water and brine, dried over sodium sulfate and concentrated in vacuo. The product was purified using silica gel chromatography (heptane/ethyl acetate = 1/4 v/v%) to give 5 g of (S)-benzyl 2-((3-chloropyrazin-2-yl)methylcarbamoyl)pyrrolidine-1-carboxylate (62.7%).
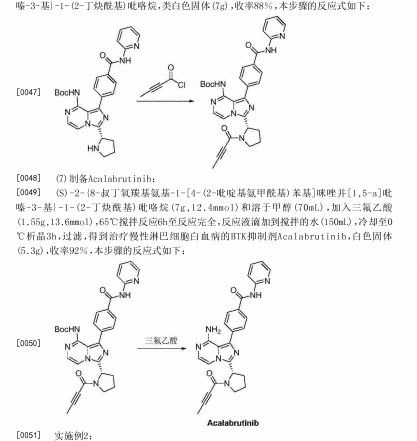
(c) (S)-Benzyl 2-(8-chloroimidazo[1 ,5-alpyrazin-3-yl)pyrrolidine-1-carboxylate
(S)-Benzyl 2-((3-chloropyrazin-2-yl)methylcarbamoyl)pyrrolidine-1-carboxylate (20.94 mmol, 7.85 g) was dissolved in acetonitrile (75 ml), 1 ,3-dimethyl-2-imidazolidinone (62.8 mmol, 6.9 ml, 7.17 g) was added and the reaction mixture was cooled to 0°C before POCI3 (84 mmol, 7.81 ml, 12.84 g) was added drop wise while the temperature remained around 5°C. The reaction mixture was refluxed at 60-65°C overnight. The reaction mixture was poured carefully in ammonium hydroxide 25% in water (250 ml)/crushed ice (500 ml) to give a yellow suspension (pH -8-9) which was stirred for 15 min until no ice was present in the suspension. Ethyl acetate was added, layers were separated and the aqueous layer was extracted with ethyl acetate (3x). The organic layers were combined and washed with brine, dried over sodium sulfate, filtered and evaporated to give 7.5 g crude product. The crude product was purified using silica gel chromatography (heptane/ethyl acetate = 1/4 v/v%) to give 6.6 g of (S)-benzyl 2-(8- chloroimidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1-carboxylate (88%).
(d) (S)-Benzyl 2-(1-bromo-8-chloroimidazo[1 ,5-alpyrazin-3-yl)pyrrolidine-1-carboxylate
N-Bromosuccinimide (24.69 mmol, 4.4 g) was added to a stirred solution of (S)-benzyl 2-(8- chloroimidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1-carboxylate (24.94 mmol, 8.9 g) in DMF (145 mL). The reaction was stirred 3 h at rt. The mixture was poored (slowly) in a stirred mixture of water (145 mL), ethyl acetate (145 mL) and brine (145 mL). The mixture was then transferred into a separating funnel and extracted. The water layer was extracted with 2×145 mL ethyl acetate. The combined organic layers were washed with 3×300 mL water, 300 mL brine, dried over sodium sulfate, filtered and evaporated. The product was purified using silica gel chromatography (ethyl acetate/heptane = 3/1 v/v%) to give 8.95 g of (S)-benzyl 2-(1-bromo-8-chloroimidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1-carboxylate (82.3%).
(e) (S)-Benzyl 2-(8-amino-1-bromoimidazo[1 ,5-alpyrazin-3-yl)pyrrolidine-1-carboxylate
(S)-Benzyl 2-(8-amino-1-bromoimidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1-carboxylate (20.54 mmol, 8.95 g) was suspended in 2-propanol (1 13 ml) in a pressure vessel. 2-propanol (50 ml) was cooled to -78°C in a pre-weighed flask (with stopper and stirring bar) and ammonia gas (646 mmol, 1 1 g) was lead through for 15 minutes. The resulting solution was added to the suspension in the pressure vessel. The vessel was closed and stirred at room temperature and a slight increase in pressure was observed. Then the suspension was heated to 1 10 °C which resulted in an increased pressure to 4.5 bar. The clear solution was stirred at 1 10 °C, 4.5 bar overnight. After 18h the pressure remained 4 bar. The reaction mixture was concentrated in vacuum, the residue was suspended in ethyl acetate and subsequent washed with water. The layers were separated and the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with water, saturated sodium chloride solution, dried over sodium sulfate and concentrated to give 7.35 g of (S)-benzyl 2-(8-amino-1-bromoimidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1- carboxylate (86%).
Intermediate 2

(S)-4-(8-Amino-3-(pyrrolidin-2-v0im^
(a) (S)-Benzyl 2-(8-amino-1-(4-(pyridin-2-ylcarbamov0
carboxylate
(S)-benzyl 2-(8-amino-1-bromoimidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1 -carboxylate (0.237 mmol, 98.5 mg) and 4-(pyridin-2-yl-aminocarbonyl)benzeneboronic acid (0.260 mmol, 63.0 mg) were suspended in a mixture of 2N aqueous potassium carbonate solution (2.37 mmol, 1 .18 mL) and dioxane (2.96 mL). Nitrogen was bubbled through the mixture, followed by the addition of 1 , 1 ‘- bis(diphenylphosphino)ferrocene palladium (ii) chloride (0.059 mmol, 47.8 mg). The reaction mixture was heated for 20 minutes at 140°C in the microwave. Water was added to the reaction mixture, followed by an extraction with ethyl acetate (2x). The combined organic layer was washed with brine, dried over magnesium sulfate and evaporated. The product was purified using silicagel and dichloromethane/methanol = 9/1 v/v% as eluent to afford 97.1 mg of (S)-benzyl 2-(8-amino-1-(4-(pyridin- 2-ylcarbamoyl)phenyl)imidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1 -carboxylate (77%).
(b) (S)-4-(8-Amino-3-(pyrrolidin-2-yl)imidazo[1 ,5-alpyrazin-1-yl)-N-(pyridin-2-yl)benzamide
To (S)-benzyl 2-(8-amino-1-(4-(pyridin-2-ylcarbamoyl)phenyl)imidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1- carboxylate (0.146 mmol, 78 mg) was added a 33% hydrobromic acid/acetic acid solution (1 1.26 mmol, 2 ml) and the mixture was left at room temperature for 1 hour. The mixture was diluted with water and extracted with dichloromethane. The aqueous phase was neutralized using 2N sodium hydroxide solution, and then extracted with dichloromethane. the organic layer was dried over magnesium sulfate, filtered and evaporated to give 34 mg of (S)-4-(8-Amino-3-(pyrrolidin-2-yl)imidazo[1 ,5-a]pyrazin-1-yl)-N- (pyridin-2-yl)benzamide (58%).
Example 6

(S)-4-(8-amino-3-n-but-2-vnoylpyrrolidin-2-vnimidazo[1 ,5-alpyrazin-1-yl)-N-(pyridin-2-yl)benzamide
This compound was prepared, in an analogues manner as described in Example 2, from the compound described in intermediate 2b and 2-butynoic acid, to afford the title compound (10.5 mg, 18.0%). Data: LCMS (B) Rt : 2.08 min; m/z 466.1 (M+H)+.
PATENT
https://www.google.com/patents/WO2016024228A1?cl=en
PATENT
CN 107056786
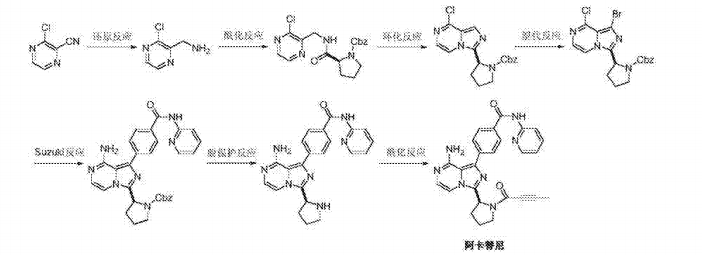
Step SI:
[0029] The pressure in the reactor was added 3-chloro-2-carboxaldehyde l-yl P ratio of (II) (0.71g, 5mmol) and dioxane (20mL), under stirring ammonia gas (I. 7g, 0 . Imol), was added 4- (pyridin-2-yl – aminocarbonyl) phenylboronic acid (III) (2.42g, lOmmol), Ming dicarbonyl acetylacetonate (0.26g, lmmol), and water 4mL. The reactor was sealed, gradually warmed to 80~90 °, the reaction 16-18 hours, TLC detection, the reaction was complete. Concentrated under reduced pressure, the residue was dissolved in dichloromethane, washed with saturated sodium bicarbonate and water successively, dried over anhydrous sodium sulfate. Concentrated to give brown oil, ethyl acetate and petroleum ether (volume ratio 1: 2) column chromatography to give an off-white solid 4- [amino (3-chloro-2-pyrazinyl) methyl] -N- (2-pyridyl) benzamide (IV) 1.38g, yield 81 · 2%; ESI-MS (m / z): 340 (m + H).
[0030] Step S2:
[0031] added in the reactor [1- (1-oxo-2-butyn-1-yl)] – L- proline (1.09g, 6mmol) and thionyl chloride (IOmL), was added dropwise 4mL of triethylamine and heated to 30 to 40 degrees, after the reaction for 2-4 hours under reduced pressure to remove excess thionyl chloride, the residue that is [I- (1- oxo-2-butyn-1-yl )] – L- proline acid chloride (V). The resulting [I- (1- oxo-2-butynyl -1_ yl)] _ L_ proline acid chloride (V) dissolved in 20mL dichloromethane burning, to a solution of 4- [amino (3-chloro -2-P ratio piperazinyl) methyl] -N- (2- pyridinyl) benzamide (IV) (1.35g, 4mmol) and triethylamine (0.6g, 6mmol) in dichloromethane (30mL) solution of in. Dropwise, warmed to 30-50 °, the reaction was stirred for 6 ~ 8 hours, TLC detection, the reaction was complete. Cooled to room temperature, washed with saturated sodium bicarbonate solution, brine and water, dried over anhydrous sodium sulfate. Concentrated to give a beige solid of 4- [1- (1-acyl-2-yne-2-yl) carboxamido (3-chloro-2-pyrazinyl) methyl] -N- (2- pyridinyl) benzamide (VI) 1.8g, yield 89.6% C3ESI-MS (m / z): 503 (m + H).
[0032] Step S3:
[0033] in a reaction flask was added 4- [I- (1- but-2-yn-acyl-2-yl) carboxamido (3-chloro-2-pyrazinyl) methyl] -N- ( 2-P ratio piperidinyl) benzamide (VI) (1 · 0g, 2mmol), phosphorus oxychloride (1 · 53g, IOmmol) and acetonitrile (25 mL), warmed to 80 ~ 100 ° with stirring, maintaining the temperature reaction 6 ~ 8 h, TLC the reaction was complete. Cooled to room temperature, the reaction solution was poured into 50mL concentration of 8% aqueous ammonia was added ethyl acetate, and the organic phase was separated, the aqueous phase was extracted twice with ethyl acetate. The combined organic phases were washed with brine and water, dried over anhydrous over sodium sulfate. Concentrated and the resulting residue with ethyl acetate and petroleum ether (volume ratio 2: 1) column chromatography to give an off-white solid 4- [8-Chloro -3- [(2S) -I- (1- oxo-2 – butyn-1-yl) -2-pyrrolidinyl] imidazo [I, 5-a] pyrazin-1-yl] -N-2- pyridinyl benzamide (VII) 0.85g, yield 87.8 %; EI-MS m / z: 485 [m + H] + square
[0034] Step S4:
[0035] The pressure reactor was added to 4- [8-Chloro -3- [(2S) -I- (1- oxo-2-butyn-1-yl) -2-pyrrolidinyl] imidazo [ I, 5-a] pyrazin – Buji] -N-2- pyridinyl benzamide (VII) (0.48g, lmmol) and isopropanol (15 mL), cooled to 0 degrees, by controlling the dose into ammonia gas (0.51g, 30mmol), the reactor is closed, warmed up to room temperature for 1 hour, and then continuously increasing the reaction temperature to 110~120 °, maintained at the reaction temperature and pressure 20~24 h, TLC the reaction was complete. Cooled to room temperature, slowly vented, and concentrated under reduced pressure, the resulting residue was dissolved with ethyl acetate, water and saturated brine, dried over anhydrous sodium sulfate. Concentrated and the resulting residue with ethyl acetate and petroleum ether (volume ratio 2: 1) column chromatography to give an off-white solid Acre imatinib ⑴ 0.40g, yield 86 · 0%; 1Η bandit R (DMS0-d6) 1.63 (m, lH), 1.97 (s, 3H), 2.02 ~2.12 (m, lH), 2 · 28~2.35 (m, 2H); 3.36~3.85 (m, 2H), 5 · 47~5.49 (m , lH), 6 · 17~6.23 (m, 2H), 7.12~7.20 (m, 2H), 7 · 73~7.86 (m, 4H), 8 · 16~8.25 (m, 3H), 8 · 41 ( dd, lH), 10.86 (s, lH); EI-MS m / z: 466 [m + H] +.
[0036] 3-chloro starting material employed in the method above relates to the present invention yl pyrazin-2-carbaldehyde (II) and 4- (pyridin-2-yl – aminocarbonyl) phenylboronic acid (III), respectively, refer to methods for their preparation Document “Tetrahedron Letters, 47 (l), 31-34; 2006” international Patent W02013010868 and method for preparing the same compound. Raw [1- (1-oxo-2-butyn-1-yl)] – L- proline acid chloride (V), in one embodiment, the compound may be made [the I-(1-oxo-known -2-yn-1-yl)] – L- proline acylation.
PATENT
US 20170224688
PATENT
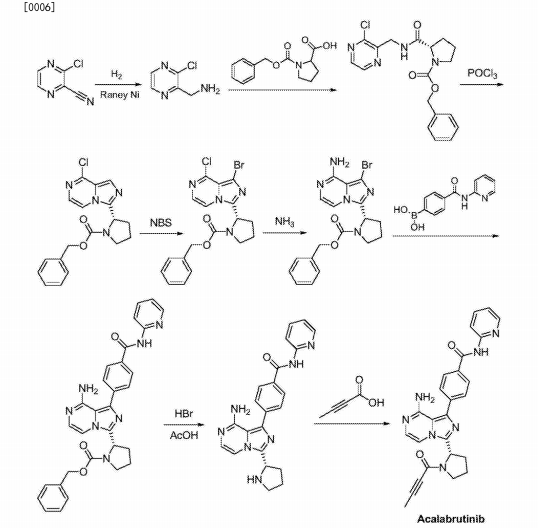

Example I
[0030] (1) Preparation of ⑸-2- (8- amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0031] (S) -2- (8- chloro-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (10g, 28mmol) was dissolved in N- methylpyrrolidone ( SOML), the mass concentration was added 28% aqueous ammonia (168mm〇l), the reaction mixture was placed in a sealed stainless steel autoclave at 85 ° C, stirring the reaction under a pressure of 2.5 atm 6h, after the completion of the reaction, was cooled to 40 ° C and delivery system pressure, slow addition of water (50 mL), cooled to 10 ° C, crystallization 3h, filtered, and recrystallized from isopropanol to give ⑸-2- (8- amino-imidazo [I, 5-a] pyrazin – 3- yl) -1-pyrrolidine-carboxylate, an off-white solid (8.5 g of), yield 90%, reaction formula of this step is as follows:

[0033] (2) Preparation of (S) -2- (8- tert-butoxycarbonyl-amino-imidazo [I, 5_a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0034] (S) -2- (8- amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (8g, 24mmol) was dissolved in dichloromethane (IOOmL) was added tert-butyl dicarbonate (5.7g, 26mmol), reaction mixture was stirred 3h at 25 ° C, after completion of the reaction, post-treatment and purification to give ⑸-2- (8- tert-butoxycarbonyl-amino-imidazole and [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate, an off-white solid (IoG), 96% yield, this step follows the reaction formula:

[0036] (3) Preparation of (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0037] (S) -2- (8- tert-butoxycarbonyl-amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (IOg, 23mmol) was dissolved in tetrahydrofuran ( 80mL), was slowly added N- bromosuccinimide (4.5g, 25mmol), the reaction mixture was 25 ° C the reaction was stirred for 4h. The mixture was then slowly added water (80 mL), cooled to -10 ° C crystallization 3h, filtered, and recrystallized from isopropanol to give (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [ I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate, an off-white solid (I I. Ig), a yield of 94.5%, the reaction formula of this step is as follows:

[0039] (4) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} 1-pyrrolidine-carboxylic acid benzyl ester:
[0040] (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (I Ig, 2lmmol ), 4- (2-pyridyl-carbamoyl) phenylboronic acid (5.7g, 23.4mmol), [1, Γ – bis (diphenylphosphino) ferrocene] dichloropalladium cesium (〇.78g, the I · lmmol), potassium carbonate (4.0g, 29mmol), N, N- dimethylformamide (120 mL) and water (50mL) added to the reaction flask, the reaction mixture was heated to 90 ° C the reaction was stirred for 20 h, the reaction solution was reduced at room temperature, was concentrated by rotary evaporation to dryness, extracted with ethyl acetate, washed with brine, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, a mixed solvent of ethyl acetate and n-hexane and recrystallized to give (S) -2- {8- tert butoxycarbonyl group -I- [4- (2-P of pyridine-ylcarbamoyl) phenyl] imidazole and sat Jie [I, 5_a] pyrazin-3-yl} -1-pyrrolidine-carboxylate, class as a white solid (10.3 g of), a yield of 76.5%, the reaction formula of this step is as follows:

[0042] (5) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} pyrrolidine:
[0043] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl } -1- [1-carboxylic acid than the burning section slightly ester (10g, 15.8mmol) was dissolved in methanol (80mL), was added cesium charcoal (0.5g), under a hydrogen pressure into 35 ° C the reaction 8h. Concentrated suction through Celite to remove the catalyst and the filtrate was rotary evaporated to dryness to afford ⑸-2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [ I, 5-a] pyrazin-3-yl} pyrrolidine as a white solid powder (7.6 g of), 96% yield, this step follows the reaction formula:

[0045] (6) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} -1- (2-butynoyl) pyrrolidine:
[0046] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl } ratio slightly burning Jie (7g, 14mmol) was dissolved in tetrahydrofuran (75 mL), with stirring, was added 2-butyne chloride (I. 7g, 16.6mmol), was added dropwise N, N- diisopropylethylamine (2.7 g, 21 mmol), the reaction mixture was 50 ° C the reaction was stirred for 8h, the reaction solution was concentrated by rotary evaporation to dryness, dilute hydrochloric acid was added was adjusted to neutral, extracted with ethyl acetate was added, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, recrystallized from methanol to give ⑸ -2_ {8-tert-butoxycarbonyl-amino -1- [4- (2-P of pyridine-ylcarbamoyl) phenyl] imidazole and sat Jie [I, 5_a] [! than 3-yl} -1 – (2_ butynoyl) pyrrolidine-white solid (7g), in 88% yield, this step follows the reaction formula:

[0048] ⑺ prepared Acalabrutinib:
[0049] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl } -1- (2-butynoyl) pyrrolidine (7g, 12.4mmol) and dissolved in methanol (70 mL), trifluoroacetic acid (1.55g, 13.6mmol), 65 ° C until the reaction was complete the reaction was stirred for 6h, the reaction was added dropwise to a stirred solution of water (150 mL), cooled to 0 ° C crystallization 3h, filtered to give the treatment of chronic lymphocytic leukemia BTK inhibitors Acalabrut inib, as a white solid (5.3 g of), 92% yield, this step is the following reaction formula:

[0051] Example 2:
[0052] (1) Preparation of ⑸-2- (8- amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0053] (S) -2- (8- chloro-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (15g, 42mmol) was dissolved in N- methylpyrrolidone ( 75 mL), aqueous ammonia (273_〇1) was added mass percent concentration of 28%, the reaction mixture was placed in a sealed stainless steel autoclave at 70 ° C, stirring the reaction under a pressure of 3 atm 8h, after the completion of the reaction, was cooled to 40 ° C and releasing the pressure in the system, slow addition of water (50 mL), cooled to 10 ° C, crystallization 3h, filtered, and recrystallized from isopropanol to give ⑸-2- (8- amino-imidazo [I, 5-a] pyrazine 3-yl) pyrrolidine-carboxylic acid benzyl ester, off-white solid (12.9 g of), yield 91% ,, this step reaction scheme in Example 1.
[0054] (2) Preparation of (S) -2- (8- tert-butoxycarbonyl-amino-imidazo [I, 5_a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0055] (S) -2- (8- amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (12g, 35.6mmol) was dissolved in chloroform (80mL), was added tert-butyl dicarbonate (7.8g, 35.6mmol), the reaction mixture was stirred for lh the reaction at 35 ° C, after completion of the reaction, post-treatment and purification to give ⑸-2- (8- tert-butoxycarbonyl-amino-imidazole and [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate, an off-white solid (14.8 g of), in 95% yield, this step is the same reaction scheme as in Example 1.
[0056] (3) Preparation of (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0057] (S) -2- (8- tert-butoxycarbonyl-amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (Hg, 32mmol) was dissolved in 1, 1,2-dichloroethane (90mL), was slowly added bromine (6g, 37.8mmol), the reaction mixture was 20 ° C the reaction was stirred for 6h. After the reaction, water was slowly added (I5mL), cooled to -5 ° C crystallization 4h, filtered and recrystallized from isopropanol to give ⑸-2- (8- tert-butoxycarbonyl-amino-1-bromo-imidazo [1, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate, an off-white solid (15.8 g), yield 95.5%, the reaction of the present step is the same formula as in Example 1.
[0058] (4) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} 1-pyrrolidine-carboxylic acid benzyl ester:
[0059] (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (15g, 29mmol) , 4- (2-pyridyl-carbamoyl) phenylboronic acid (34 · 7mmol 8 · 4g,), tetrakis (triphenylphosphine) palladium (0 · 84g, 0.73mmol), sodium carbonate (6.9g, 65mmol), tetrahydrofuran (IOOmL) and water (40 mL) was added a reaction flask, the reaction mixture was heated to 80 ° C the reaction was stirred for 24h, the reaction was cooled to room temperature, and concentrated by rotary evaporation to dryness, extracted with ethyl acetate, washed with brine, dried over magnesium sulfate, concentrated by rotary evaporation to dryness, a mixed solvent of ethyl acetate and n-hexane and recrystallized to give ⑸-2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazole and [I, 5-a] pyrazin-3-yl} -1-pyrrolidine-carboxylate, an off-white solid (14.4g), 78% yield, this step is the same reaction scheme as in Example 1.
[0060] (5) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} pyrrolidine:
[0061] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-yl _3- it is slightly burned} -1-carboxylic acid ester section (14g, 22mmol) dissolved in isopropanol (85mL), was added Raney nickel (0.5g), under a hydrogen pressure into the reaction 60 ° C 12h. Concentrated suction through Celite to remove the catalyst and the filtrate was rotary evaporated to dryness to afford ⑸-2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [ I, 5-a] pyrazin-3-yl} pyrrolidine as a white solid powder (10.4 g of), 94% yield, this step is the same reaction scheme as in Example 1.
[0062] (6) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} -1- (2-butynoyl) pyrrolidine:
[0063] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl } pyrrolidine (10g, 20mmo 1) was dissolved in N, N- dimethylformamide (SOML), with stirring, was added 2-butyne chloride (3. lg, 30mmol), dropwise addition of triethylamine (2.2g, 22mmol ), the reaction mixture was 60 ° C the reaction was stirred for 4h, the reaction solution was concentrated by rotary evaporation to dryness, dilute hydrochloric acid was added was adjusted to neutral, extracted with ethyl acetate was added, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, and recrystallized from methanol to give ⑸- 2- {8-tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl} -l- (2- butynoyl) pyrrolidine-white solid (10.2 g of), a yield of 90.2%, the same reaction scheme of the present embodiment step 1〇
[0064] ⑺ prepared Acalabrutinib:
[0065] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl } -1- (2-butynoyl) pyrrolidine (IOg, 17.7mmol) was dissolved in ethanol, and (IOOmL), trifluoroacetic acid (2.6g, 23mmol), 50 ° C with stirring until the reaction was complete IOh reaction, the reaction solution was added dropwise to a stirred solution of water (70 mL), cooled to 0 ° C crystallization 3h, filtered to give the treatment of chronic lymphocytic leukemia BTK inhibitors AcaIabrut inib, as a white solid (7.5 g of), yield 91%, reaction of this step formula same as in Example 1.
[0066] Example 3:
[0067] (1) Preparation of ⑸-2- (8- amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0068] (S) -2- (8- chloro-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (4.5g, 12.6mmol) was dissolved in N- methyl pyrrolidinone (70 mL), was added mass percent concentration of 28% aqueous ammonia (69.4 mmol), the reaction mixture was placed in the autoclave 90 ° C, the reaction was stirred under atmospheric pressure of 4h, after the completion of the reaction, it was cooled to 35 ° C a sealed stainless steel reactor and releasing the pressure in the system, slow addition of water (50 mL), cooled to 10 ° C, crystallization 3h, filtered, and recrystallized from isopropanol to give ⑸-2- (8- amino-imidazo [I, 5-a] pyrazine 3-yl) pyrrolidine-carboxylic acid benzyl ester, off-white solid (3.9 g of), 92% yield, this step is the same reaction scheme as in Example 1.
[0069] (2) Preparation of (S) -2- (8- tert-butoxycarbonyl-amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0070] (S) -2- (8- amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester (3 · 5g, 10 · 4mmol) was dissolved in 1, 4- dioxane (50 mL), was added tert-butyl dicarbonate (2.7g, 12.4mmol), the reaction mixture was stirred at 10 ° C the reaction 6h, after the completion of the reaction, workup and purification, to give (S) 2- (8-tert-butoxycarbonyl-amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate, an off-white solid (4.3 g of), in 95% yield, according to the present step reaction scheme in Example 1.
[0071] (3) Preparation of (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0072] (S) -2- (8- tert-butoxycarbonyl-amino-imidazo [l, 5_a] pyrazin-3-yl) -1_ pyrrolidine-carboxylate (4g, 9.6mmol) was dissolved in toluene (50 mL ), was slowly added N- bromosuccinimide (I. 8g, 10. lmmol), the reaction mixture was 35 ° C the reaction was stirred for 2h. The mixture was then slowly added water (25 mL), cooled to -10 ° C crystallization 3h, filtered, and recrystallized from isopropanol to give (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [ I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate, an off-white solid (4.7 g), 94% yield, this step is the same reaction scheme as in Example 1.
[0073] (4) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} 1-pyrrolidine-carboxylic acid benzyl ester:
[0074] (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (4g, 7 · 7mmol), 4_ (2- piperidinyl than Jie carbamoyl) phenylboronic acid (2 · 4g, IOmmol), bis (triphenylphosphine) dichloride Leba (0.41g, 0.58mmol), potassium phosphate (I. 9g, 8.9mmol), methyl tert-butyl ether (IOOmL) and water (40 mL) was added a reaction flask, the reaction mixture was heated to 100 ° C the reaction was stirred for 12h, the reaction was cooled to room temperature, and concentrated by rotary evaporation to dryness, was added acetic acid extracted with ethyl, brine, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, a mixed solvent of ethyl acetate and n-hexane and recrystallized to give ⑸-2- {8- tert-butoxycarbonyl-amino-1- [4- (2 – pyridin-ylcarbamoyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl} -1-pyrrolidine-carboxylate, an off-white solid (3.9 g of), in 79% yield, this step the reaction scheme in Example 1.
[0075] (5) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5_a] pyrazin-3 -} pyrrolidine:
[0076] (S) -2- {8- tert-butoxycarbonyl group -I- [4- (2- carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl } -1 Jie section than slightly burning acid ester (3.5g, 5.5mmol) was dissolved in ethanol (50mL), was added cesium charcoal (0.2g), under a hydrogen pressure into 45 ° C the reaction 6h. Concentrated suction through Celite to remove the catalyst and the filtrate was rotary evaporated to dryness to afford ⑸-2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [ I, 5-a] pyrazin-3-yl} pyrrolidine as a white solid powder (2.6 g of), in 95% yield, this step is the same reaction scheme as in Example 1.
[0077] (6) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} -1- (2-butynoyl) pyrrolidine:
[0078] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl } ratio slightly burning Jie (2.5g, 5mmol) was dissolved in toluene (50 mL), with stirring, was added 2-butyne chloride (0.62g, 6mmol), was added dropwise N, N- dimethylaniline (Ig, 8.5mmo 1), The reaction mixture was 40 ° C the reaction was stirred for 12h, the reaction solution was concentrated by rotary evaporation to dryness, dilute hydrochloric acid was added was adjusted to neutral, extracted with ethyl acetate was added, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, and recrystallized from methanol to give ⑸-2- {8-tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl} -1- (2-butyn acyl) pyrrolidine-white solid (2.5g), 88% yield, this step is the same reaction scheme as in Example 1.
[0079] ⑺ prepared Acalabrutinib:
[0080] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-yl _3_ } -1- (2-block group) ratio slightly burning Jie (2.5g, 4.4mmol) was dissolved in dichloromethane and burned (IOmL), two gas was added acetic acid (0.76g, 6.6mmol), 80 ° C The reaction was stirred 4h until the reaction was complete, the reaction was added dropwise to a stirred solution of water (25 mL), cooled to 0 ° C crystallization 3h, filtered to give the treatment of chronic lymphocytic leukemia BTK inhibitors AcaIabrut inib, as a white solid (1.8 g of), the yield of 89%, this step is the same reaction scheme as in Example 1.
PATENT
US 20170035881
References
- Jump up^ “WHO Drug Information – recommended INN” (PDF). WHO Drug Information. World Health Oorganisation. Retrieved 24 December 2015.
- ^ Jump up to:a b c d Byrd; et al. (2015). “Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia”. doi:10.1056/NEJMoa1509981.
- ^ Jump up to:a b c d e f g h i Wu, Jingjing; Zhang, Mingzhi; Liu, Delong (2016-01-01). “Acalabrutinib (ACP-196): a selective second-generation BTK inhibitor”. Journal of Hematology & Oncology. 9: 21. doi:10.1186/s13045-016-0250-9. ISSN 1756-8722. PMC 4784459
 . PMID 26957112.
. PMID 26957112. - Jump up^ “AstraZeneca to buy Acerta for blood cancer drug”. www.rsc.org. Chemistry World – Royal Society of Chemistry. Retrieved 24 December 2015.
- Jump up^ Wu, Jingjing; Zhang, Mingzhi; Liu, Delong (2016-03-09). “Acalabrutinib (ACP-196): a selective second-generation BTK inhibitor”. Journal of Hematology & Oncology. 9 (1). doi:10.1186/s13045-016-0250-9. ISSN 1756-8722. PMC 4784459 . PMID 26957112.
- ^ Jump up to:a b c Harrington, Bonnie K.; Gardner, Heather L.; Izumi, Raquel; Hamdy, Ahmed; Rothbaum, Wayne; Coombes, Kevin R.; Covey, Todd; Kaptein, Allard; Gulrajani, Michael (2016-07-19). “Preclinical Evaluation of the Novel BTK Inhibitor Acalabrutinib in Canine Models of B-Cell Non-Hodgkin Lymphoma”. PLOS ONE. 11 (7): e0159607. doi:10.1371/journal.pone.0159607. ISSN 1932-6203. PMC 4951150 . PMID 27434128.
- ^ Jump up to:a b Acerta Pharma Announces Study Published in New England Journal of Medicine Demonstrates Acalabrutinib (ACP-196) Shows Marked Activity in Relapsed Chronic Lymphocytic Leukemia
- Jump up^ 21 studies found for: ACP-196
- Jump up^ “Acerta Investor Conference Call – 17 December 2015” (PDF). www.astrazeneca.com. Retrieved 2016-11-20.
- Jump up^ “Public summary of opinion on orphan designation” (PDF). European Medicines Agency. 2016-04-27. Retrieved 2016-11-20.
- Jump up^ “azn201602256k.htm”. www.sec.gov. Retrieved 2016-11-21.
- Jump up^ House, SA Editor Douglas W. (2016-02-25). “AstraZeneca and Acerta Pharma’s acalabrutinib tagged an Orphan Drug in Europe for three indications”. Seeking Alpha. Retrieved 2016-11-21.
- Jump up^ “Acerta Pharma B.V. – Company Profile – BioCentury”. www.biocentury.com. Retrieved 2016-11-12.
- Jump up^ “Log in to CB Insights”. www.cbinsights.com. Retrieved 2016-11-12.
- Jump up^ “This is The Most Valuable Startup You’ve Never Heard Of”. Fortune. 2015-12-17. Retrieved 2016-11-12.
- Jump up^ Walker, Ian; Roland, Denise (2015-12-17). “AstraZeneca to Buy Stake in Acerta Pharma”. Wall Street Journal. ISSN 0099-9660. Retrieved 2016-11-19.
- Jump up^ HAMDY, Ahmed; Rothbaum, Wayne; IZUMI, Raquel; Lannutti, Brian; Covey, Todd; ULRICH, Roger; Johnson, Dave; Barf, Tjeerd; Kaptein, Allard (Nov 26, 2015), Btk inhibitor for the treatment of chronic lymphocytic and small lymphocytic leukemia, retrieved 2016-11-19
- Jump up^ HAMDY, Ahmed; Rothbaum, Wayne; IZUMI, Raquel; Lannutti, Brian; Covey, Todd; ULRICH, Roger; Johnson, Dave; Barf, Tjeerd; Kaptein, Allard (Jun 11, 2015), Therapeutic combination of a pi3k inhibitor and a btk inhibitor, retrieved 2016-11-19
- ^ Jump up to:a b IZUMI, Raquel; SALVA, Francisco; HAMDY, Ahmed (Feb 4, 2016), Methods of blocking the cxcr-4/sdf-1 signaling pathway with inhibitors of bruton’s tyrosine kinase, retrieved 2016-11-19
- Jump up^ HAMDY, Ahmed; Rothbaum, Wayne; IZUMI, Raquel; Lannutti, Brian; Covey, Todd; ULRICH, Roger; Johnson, Dave; Barf, Tjeerd; Kaptein, Allard (Aug 4, 2016), Therapeutic combinations of a btk inhibitor, a pi3k inhibitor and/or a jak-2 inhibitor, retrieved 2016-11-19
- Jump up^ Lannutti, Brian; Covey, Todd; Kaptein, Allard; Johnson, David; STAMATIS, Jay; Krejsa, Cecile M.; Slatter, John Gregory (Feb 11, 2016), Methods of treating cancers, immune and autoimmune diseases, and inflammatory diseases based on btk occupancy and btk resynthesis rate, retrieved 2016-11-19
- Jump up^ HAMDY, Ahmed; Rothbaum, Wayne; IZUMI, Raquel; Lannutti, Brian; Covey, Todd; ULRICH, Roger; Johnson, Dave; Barf, Tjeerd; Kaptein, Allard (Feb 18, 2016), Btk inhibitors to treat solid tumors through modulation of the tumor microenvironment, retrieved 2016-11-19
- Jump up^ HAMDY, Ahmed; Rothbaum, Wayne; IZUMI, Raquel; Lannutti, Brian; Covey, Todd; ULRICH, Roger; Johnson, Dave; Barf, Tjeerd; Kaptein, Allard (Feb 18, 2016), Therapeutic combinations of a btk inhibitor, a pi3k inhibitor, a jak-2 inhibitor, and/or a bcl-2 inhibitor, retrieved 2016-11-19
- Jump up^ HAMDY, Ahmed; Rothbaum, Wayne; IZUMI, Raquel; Lannutti, Brian; Covey, Todd; ULRICH, Roger; Johnson, Dave; Barf, Tjeerd; Kaptein, Allard (Feb 18, 2016), Therapeutic combinations of a btk inhibitor, a pi3k inhibitor, a jak-2 inhibitor, a pd-1 inhibitor and/or a pd-l1 inhibitor, retrieved 2016-11-19
- Jump up^ HAMDY, Ahmed; Rothbaum, Wayne; IZUMI, Raquel; Lannutti, Brian; Covey, Todd; ULRICH, Roger; Johnson, Dave; Barf, Tjeerd; Kaptein, Allard (Feb 18, 2016), Therapeutic combinations of a btk inhibitor, a pi3k inhibitor, a jak-2 inhibitor and/or a cdk 4/6 inhibitor, retrieved 2016-11-19
- Jump up^ HAMDY, Ahmed; Rothbaum, Wayne; IZUMI, Raquel; Lannutti, Brian; Covey, Todd; ULRICH, Roger; Johnson, Dave; Barf, Tjeerd; Kaptein, Allard (Jul 28, 2016), Compositions and methods for treatment of chronic lymphocytic leukemia and small lymphocytic leukemia using a btk inhibitor, retrieved 2016-11-19
- Jump up^ HAMDY, Ahmed; Rothbaum, Wayne; IZUMI, Raquel; Lannutti, Brian; Covey, Todd; ULRICH, Roger; Johnson, Dave; Barf, Tjeerd; Kaptein, Allard (Aug 18, 2016), Therapeutic combinations of a btk inhibitor, a pi3k inhibitor, a jak-2 inhibitor, a pd-1 inhibitor, and/or a pd-l1 inhibitor, retrieved 2016-11-19
- Jump up^ Barf, Tjeerd A.; Jans, Christian Gerardus Johannes Maria; Man, Petrus Antonius De Adrianus; Oubrie, Arthur A.; Raaijmakers, Hans C. A.; Rewinkel, Johannes Bernardus Maria; Sterrenburg, Jan-Gerard; Wijkmans, Jacobus C. H. M. (5 June 2014), United States Patent Application: 0140155385 – 4-IMIDAZOPYRIDAZIN-1-YL-BENZAMIDES AND 4-IMIDAZOTRIAZIN-1-YL-BENZAMIDES AS BTK INHIBITORS, retrieved 2016-11-19
ADDITIONAL INFORMATION
Acalabrutinib is a potent and selective BTK (Bruton’s tyrosine kinase) inhibitor. BTK is a cytoplasmic, non-receptor tyrosine kinase that transmits signals from a variety of cell-surface molecules, including the B-cell receptor (BCR) and tissue homing receptors. Genetic BTK deletion causes B-cell immunodeficiency in humans and mice, making this kinase an attractive therapeutic target for B-cell disorders. BTK inhibitors targeting B cell receptor signaling and other survival mechanism showed great promise for the treatment of chronic lymphocytic leukemia (CLL)s holds great promise.
As of 2015 it is in late stage clinical trials for relapsed chronic lymphocytic leukemia. Interim results are encouraging : 95% overall response rate. It is also in another 20 clinical trials (alone and in combination) for various cancers.
REFERENCES
1: Maly J, Blachly JS. Chronic Lymphocytic Leukemia: Exploiting Vulnerabilities with Targeted Agents. Curr Hematol Malig Rep. 2016 Feb 11. [Epub ahead of print] PubMed PMID: 26893063.
2: Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, Chaves J, Wierda WG, Awan FT, Brown JR, Hillmen P, Stephens DM, Ghia P, Barrientos JC, Pagel JM, Woyach J, Johnson D, Huang J, Wang X, Kaptein A, Lannutti BJ, Covey T, Fardis M, McGreivy J, Hamdy A, Rothbaum W, Izumi R, Diacovo TG, Johnson AJ, Furman RR. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016 Jan 28;374(4):323-32. doi: 10.1056/NEJMoa1509981. Epub 2015 Dec 7. PubMed PMID: 26641137.
 |
|
| Identifiers | |
|---|---|
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C26H23N7O2 |
| Molar mass | 465.507 g/mol |
| 3D model (JSmol) | |
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 9290504 |
| Expiration | Jul 11, 2032 |
| Applicant | ASTRAZENECA |
| Drug Application | N210259 (Prescription Drug: CALQUENCE. Ingredients: ACALABRUTINIB) |
////////////Acalabrutinib, rINN, ACP-196, fda 2017, Акалабрутиниб , أكالابروتينيب , 阿可替尼 , Orphan Drug, breakthrough therapy designation, Lymphoma, mantle cell, ACERTA PHARMA
CC#CC(=O)N1CCC[C@H]1c2nc(c3n2ccnc3N)c4ccc(cc4)C(=O)Nc5ccccn5
CC#CC(=O)N1CCCC1C2=NC(=C3N2C=CN=C3N)C4=CC=C(C=C4)C(=O)NC5=CC=CC=N5