
Alcaftadine
NEW PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017211246&redirectedID=true
WO-2017211246, SHENZHEN TARGETRX, INC.
SUBSTITUTED FUSED IMIDAZOLE CYCLIC COMPOUND AND PHARMACEUTICAL COMPOSITION THEREOF
WANG, Yihan; (CN).
XING, Qingfeng; (CN)
Novel deuterated analogs of substituted fused imidazole cyclic compounds, particularly alcaftadine are histamine H1-receptor antagonists and mast cell stabilizers, useful for treating allergy and nasal congestion.

The present invention relates to a substituted fused imidazole cyclic compound and a composition containing said compound and application thereof. Specifically disclosed is the fused imidazole cyclic compound represented by formula (I), or a pharmaceutical composition of its crystalline form, pharmaceutically acceptable salt, prodrug, stereoisomer, hydrate, or solvate. The compound of the present invention may be used as a histamine H1-receptor antagonist and mast-cell stabilizer, and is capable of inhibiting mast-cell release of histamine and preventing histamine function, thereby reducing allergic reaction
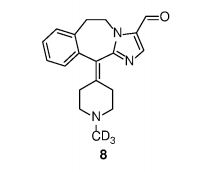
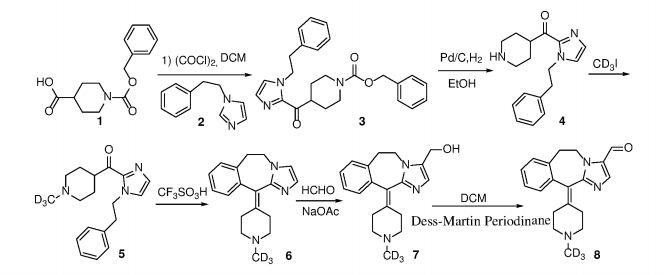
Example 1 Preparation of 6,11-dihydro -11- (1- (d3- methyl) piperidin-4-ylidene) -5H- imidazo [2,1-b] [3] benzazepine  – 3- aldehyde (compound 8)
– 3- aldehyde (compound 8)
Step 1. Synthesis of compound 3.
N-benzyloxycarbonylpiperidine-4-carboxylic acid (2.63 g, 10 mmol) was dissolved in 20 mL of dichloromethane, 6 mL of oxalyl chloride and 1 drop of DMF were added and the mixture was reacted at room temperature for 2 hours under nitrogen. The reaction mixture was concentrated to dryness under reduced pressure, dissolved in 20 mL of acetonitrile, and added with triethylamine (4.1 mL, 30 mmol) in an ice bath and stirred for 3 minutes. A solution of 1-phenethyl-1H-imidazole (2.06 g, 12 mmol) in 5 mL of acetonitrile was slowly added dropwise and the reaction was allowed to warm to room temperature overnight after the addition was completed. The reaction was completed, concentrated to dryness, 30 mL of ethyl acetate and 20 mL of water were added and the mixture was stirred for 5 minutes. The layers were separated and the aqueous phase was extracted with ethyl acetate. The combined organic phases were dried over anhydrous sodium sulfate and concentrated to give 3.34 g of a colorless oil, benzyl-4- (1-phenethyl-1H-imidazole-2-formyl) piperidine-1-carboxylate (Compound 3) was obtained in a yield of 80%. ESI-MS: 418 [M ++ 1].
Step 2. Synthesis of compound 4.
Benzyl-4- (1-phenylethyl-1H-imidazole-2-formyl) piperidine-1-carboxylate (3.34 g, 8 mmol) was dissolved in 30 mL of absolute ethanol and 300 mg of 10% palladium on carbon , Hydrogen was substituted three times and stirred overnight at room temperature under a hydrogen atmosphere of 1 atmosphere. After completion of the reaction, the palladium carbon was filtered off and the filtrate was concentrated. 2.04 g of (1-phenethyl-1H-imidazol-2-yl) (piperidin-4-yl) methanone (Compound 4) 90%. ESI-MS: 284 [M ++ 1].
Step 3. Synthesis of compound 5.
(Piperidin-4-yl) methanone (2.04 g, 7.2 mmol) was dissolved in 10 mL of DMF and potassium carbonate (1.98 g, 14.4 mmol) The solution was cooled to -15 ° C and deuterated methyl iodide (1.02 g, 7.2 mmol) was slowly added dropwise under the protection of nitrogen. After the addition was completed, the mixture was stirred at room temperature for 0.5 hour. The mixture was extracted with ethyl acetate and extracted with ethyl acetate. The organic phase was washed once with 20 mL of water and 20 mL of saturated brine, dried over anhydrous sodium sulfate, concentrated and separated on a silica gel column (1- (methyl-d3) piperidine (1-phenethyl-1H-imidazol-2-yl) methanone (Compound 5) was obtained in an amount of 70%. 1 H NMR (300 MHz, CDCl 3 ) δ 7.23 (d, J = 2.0Hz, 1H), 7.06 (td, J = 4.2,3.8,1.7Hz, 3H), 6.86 (d, J = 1.0Hz, 1H) (Dd, J = 10.2, 5.8 Hz, 2H), 3.09 (t, J = 7.2 Hz, 2H) J = 7.2 Hz, 2H), 2.85-2.65 (m, 2H), 2.15 (td, J = 7.5, 3.9 Hz, 4H); ESI-MS: 301 [M ++ l ].
Step 4. Synthesis of Compound 6.
(1-phenethyl-1H-imidazol-2-yl) methanone (1.5 g, 5.1 mmol) was placed in a reaction flask and the mixture was purged with nitrogen three times , 7mL trifluoromethanesulfonic acid was added dropwise, the reaction was warmed to 110 ° C overnight. Cooled to room temperature, the reaction solution was poured into 30mL ice water, 50% sodium hydroxide solution was added dropwise to adjust the pH = 10-11, extracted with dichloromethane, the organic phase was washed once with 20mL of water and 20mL of saturated brine, Dried over sodium sulfate, concentrated and separated by silica gel column to obtain 0.85 g of compound 6, yield 60%. 1 H NMR (300 MHz, CDCl 3 ) δ 7.28 (d, J = 4.4 Hz, 2H), 7.23 (d, J = 5.0 Hz, 1H), 7.13 (d, J = 7.0 Hz, 1H), 7.02 (D, J = 1.3Hz, 1H), 4.38 (dt, J = 12.7, 3.9Hz, 1H), 4.02 (td, J = 13.3,3.1Hz, 1H), 3.59 -3.34 (m, 3H), 3.21 (s, 2H), 3.04-2.87 (m, 3H), 2.78-2.63 (m, 2H). ESI-MS: 283 [M ++ l ].
Step 5. Synthesis of compound 7.
Compound 6 (850 mg, 3 mmol) was placed in a reaction flask, followed by the addition of 0.5 mL of acetic acid, 5 mL of 37% formaldehyde and sodium acetate (87 mg, 1.1 mmol) and warming to 100 ° C overnight. After the reaction was cooled to room temperature completely, 30 mL of methylene chloride was added to the reaction solution, 50% sodium hydroxide solution was added dropwise to adjust pH = 11-12, stirred for 0.5 hour, and the layers were separated and the organic phase was washed with 10 mL of saturated saline , Dried over anhydrous sodium sulfate, concentrated and separated on a silica gel column to give the compound 7 340 mg, yield 36%. ESI-MS: 313 [M ++ 1].
Step 6. Synthesis of Compound 8.
Compound 7 (340 mg, 1.1 mmol) was dissolved in 20 mL of dichloromethane and 4-dimethylaminopyridine (DMAP, 13 mg, 0.11 mmol) and Dess-Martin Periodinane 1.3 mmol) and reacted at room temperature for 3 hours. Join 20mL saturated sodium bicarbonate solution and 20mL dichloromethane, stirred for 5 minutes, filtered and the filtrate was separated. The organic phase was washed with saturated brine, dried over anhydrous sodium sulfate and concentrated. The compound 8 270mg was obtained by silica gel column, and the yield was 80%. 1 H NMR (300 MHz, CDCl 3 ) δ 9.64 (s, 1H), 7.76 (s, 1H), 7.34-7.26 (m, 3H), 7.16 (d, J = 6.7 Hz, 1H), 4.74 J = 14.5, 3.9 Hz, 1H), 4.31 (td, J = 14.1, 3.2 Hz, 1H), 3.53 (td, 3.03-2.89 (m, 4H), 2.64-2.81 (m, 4H); ESI-MS: 311 [M ++ l ].
//////////////














