














Alpha lipoic acid
(+)-Thioctic acid
- Molecular FormulaC8H14O2S2
- Average mass206.326 Da
Lipoic acid (LA), also known as α-lipoic acid, alpha-lipoic acid (ALA) and thioctic acid, is an organosulfur compound derived from caprylic acid (octanoic acid).[3] ALA is made in animals normally, and is essential for aerobic metabolism. It is also manufactured and is available as a dietary supplement in some countries where it is marketed as an antioxidant, and is available as a pharmaceutical drug in other countries.[3]
Physical and chemical properties
Lipoic acid (LA), also known as α-lipoic acid,[3][4] alpha-lipoic acid (ALA), and thioctic acid[5] is an organosulfur compound derived from octanoic acid.[3] LA contains two sulfur atoms (at C6 and C8) connected by a disulfide bond and is thus considered to be oxidized although either sulfur atom can exist in higher oxidation states.[3]
The carbon atom at C6 is chiral and the molecule exists as two enantiomers (R)-(+)-lipoic acid (RLA) and (S)-(-)-lipoic acid (SLA) and as a racemic mixture (R/S)-lipoic acid (R/S-LA).
LA appears physically as a yellow solid and structurally contains a terminal carboxylic acid and a terminal dithiolane ring.
For use in dietary supplement materials and compounding pharmacies, the USP has established an official monograph for R/S-LA.[6][7]
Biological function
“Lipoate” is the conjugate base of lipoic acid, and the most prevalent form of LA under physiological conditions.[3] Most endogenously produced RLA are not “free” because octanoic acid, the precursor to RLA, is bound to the enzyme complexes prior to enzymatic insertion of the sulfur atoms. As a cofactor, RLA is covalently attached by an amide bond to a terminal lysine residue of the enzyme’s lipoyl domains. One of the most studied roles of RLA is as a cofactor of the pyruvate dehydrogenase complex (PDC or PDHC), though it is a cofactor in other enzymatic systems as well (described below).[3]
Only the (R)-(+)-enantiomer (RLA) exists in nature and is essential for aerobic metabolism because RLA is an essential cofactor of many enzyme complexes.[3]
Biosynthesis and attachment
The precursor to lipoic acid, octanoic acid, is made via fatty acid biosynthesis in the form of octanoyl-acyl carrier protein.[3] In eukaryotes, a second fatty acid biosynthetic pathway in mitochondria is used for this purpose.[3] The octanoate is transferred as a thioester of acyl carrier protein from fatty acid biosynthesis to an amide of the lipoyl domain protein by an enzyme called an octanoyltransferase.[3] Two hydrogens of octanoate are replaced with sulfur groups via a radical SAM mechanism, by lipoyl synthase.[3] As a result, lipoic acid is synthesized attached to proteins and no free lipoic acid is produced. Lipoic acid can be removed whenever proteins are degraded and by action of the enzyme lipoamidase.[8] Free lipoate can be used by some organisms as an enzyme called lipoate protein ligase that attaches it covalently to the correct protein. The ligase activity of this enzyme requires ATP.[9]
Cellular transport
Along with sodium and the vitamins biotin (B7) and pantothenic acid (B5), lipoic acid enters cells through the SMVT (sodium-dependent multivitamin transporter). Each of the compounds transported by the SMVT is competitive with the others. For example research has shown that increasing intake of lipoic acid[10] or pantothenic acid[11] reduces the uptake of biotin and/or the activities of biotin-dependent enzymes.
Enzymatic activity
Lipoic acid is a cofactor for at least five enzyme systems.[3] Two of these are in the citric acid cycle through which many organisms turn nutrients into energy. Lipoylated enzymes have lipoic acid attached to them covalently. The lipoyl group transfers acyl groups in 2-oxoacid dehydrogenase complexes, and methylamine group in the glycine cleavage complex or glycine dehydrogenase.[3]
2-Oxoacid dehydrogenase transfer reactions occur by a similar mechanism in:
- the pyruvate dehydrogenase complex
- the α-ketoglutarate dehydrogenase or 2-oxoglutarate dehydrogenase complex
- the branched-chain oxoacid dehydrogenase (BCDH) complex
- the acetoin dehydrogenase complex.
The most-studied of these is the pyruvate dehydrogenase complex.[3] These complexes have three central subunits: E1-3, which are the decarboxylase, lipoyl transferase, and dihydrolipoamide dehydrogenase, respectively. These complexes have a central E2 core and the other subunits surround this core to form the complex. In the gap between these two subunits, the lipoyl domain ferries intermediates between the active sites.[3] The lipoyl domain itself is attached by a flexible linker to the E2 core and the number of lipoyl domains varies from one to three for a given organism. The number of domains has been experimentally varied and seems to have little effect on growth until over nine are added, although more than three decreased activity of the complex.[12]
Lipoic acid serves as co-factor to the acetoin dehydrogenase complex catalyzing the conversion of acetoin (3-hydroxy-2-butanone) to acetaldehyde and acetyl coenzyme A.[3]
The glycine cleavage system differs from the other complexes, and has a different nomenclature.[3] In this system, the H protein is a free lipoyl domain with additional helices, the L protein is a dihydrolipoamide dehydrogenase, the P protein is the decarboxylase, and the T protein transfers the methylamine from lipoate to tetrahydrofolate (THF) yielding methylene-THF and ammonia. Methylene-THF is then used by serine hydroxymethyltransferase to synthesize serine from glycine. This system is part of plant photorespiration.[13]
Biological sources and degradation
Lipoic acid is present in many foods in which it is bound to lysine in proteins,[3] but slightly more so in kidney, heart, liver, spinach, broccoli, and yeast extract.[14] Naturally occurring lipoic acid is always covalently bound and not readily available from dietary sources.[3] In addition, the amount of lipoic acid present in dietary sources is low. For instance, the purification of lipoic acid to determine its structure used an estimated 10 tons of liver residue, which yielded 30 mg of lipoic acid.[15] As a result, all lipoic acid available as a supplement is chemically synthesized.
Baseline levels (prior to supplementation) of RLA and R-DHLA have not been detected in human plasma.[16] RLA has been detected at 12.3−43.1 ng/mL following acid hydrolysis, which releases protein-bound lipoic acid. Enzymatic hydrolysis of protein bound lipoic acid released 1.4−11.6 ng/mL and <1-38.2 ng/mL using subtilisin and alcalase, respectively.[17][18][19]
Digestive proteolytic enzymes cleave the R-lipoyllysine residue from the mitochondrial enzyme complexes derived from food but are unable to cleave the lipoic acid-L–lysine amide bond.[20] Both synthetic lipoamide and (R)-lipoyl-L-lysine are rapidly cleaved by serum lipoamidases, which release free (R)-lipoic acid and either L-lysine or ammonia.[3] Little is known about the degradation and utilization of aliphatic sulfides such as lipoic acid, except for cysteine.[3]
Lipoic acid is metabolized in a variety of ways when given as a dietary supplement in mammals.[3][21] Degradation to tetranorlipoic acid, oxidation of one or both of the sulfur atoms to the sulfoxide, and S-methylation of the sulfide were observed. Conjugation of unmodified lipoic acid to glycine was detected especially in mice.[21] Degradation of lipoic acid is similar in humans, although it is not clear if the sulfur atoms become significantly oxidized.[3][22] Apparently mammals are not capable of utilizing lipoic acid as a sulfur source.
Chemical synthesis
-Lipoic_acid_molecular_structure.png)
(R)-Lipoic acid (RLA, top) and (S)-lipoic acid (SLA, down). A 1:1 mixture (racemate) of (R)- and (S)-lipoic acid is called (RS)-lipoic acid or (±)-lipoic acid (R/S-LA).
SLA did not exist prior to chemical synthesis in 1952.[23][24] SLA is produced in equal amounts with RLA during achiral manufacturing processes. The racemic form was more widely used clinically in Europe and Japan in the 1950s to 1960s despite the early recognition that the various forms of LA are not bioequivalent.[25] The first synthetic procedures appeared for RLA and SLA in the mid-1950s.[26][27][28][29] Advances in chiral chemistry led to more efficient technologies for manufacturing the single enantiomers by both classical resolution and asymmetric synthesis and the demand for RLA also grew at this time. In the 21st century, R/S-LA, RLA and SLA with high chemical and/or optical purities are available in industrial quantities. At the current time, most of the world supply of R/S-LA and RLA is manufactured in China and smaller amounts in Italy, Germany, and Japan. RLA is produced by modifications of a process first described by Georg Lang in a Ph.D. thesis and later patented by DeGussa.[30][31] Although RLA is favored nutritionally due to its “vitamin-like” role in metabolism, both RLA and R/S-LA are widely available as dietary supplements. Both stereospecific and non-stereospecific reactions are known to occur in vivo and contribute to the mechanisms of action, but evidence to date indicates RLA may be the eutomer (the nutritionally and therapeutically preferred form).[32][33]
Pharmacology
Pharmacokinetics
A 2007 human pharmacokinetic study of sodium RLA demonstrated the maximum concentration in plasma and bioavailability are significantly greater than the free acid form, and rivals plasma levels achieved by intravenous administration of the free acid form.[34] Additionally, high plasma levels comparable to those in animal models where Nrf2 was activated were achieved.[34]
The various forms of LA are not bioequivalent.[25][non-primary source needed] Very few studies compare individual enantiomers with racemic lipoic acid. It is unclear if twice as much racemic lipoic acid can replace RLA.[34]
The toxic dose of LA in cats is much lower than that in humans or dogs and produces hepatocellular toxicity.[35]
Pharmacodynamics
The mechanism and action of lipoic acid when supplied externally to an organism is controversial. Lipoic acid in a cell seems primarily to induce the oxidative stress response rather than directly scavenge free radicals. This effect is specific for RLA.[4] Despite the strongly reducing milieu, LA has been detected intracellularly in both oxidized and reduced forms.[36] LA is able to scavenge reactive oxygen and reactive nitrogen species in a biochemical assay due to long incubation times, but there is little evidence this occurs within a cell or that radical scavenging contributes to the primary mechanisms of action of LA.[4][37] The relatively good scavenging activity of LA toward hypochlorous acid (a bactericidal produced by neutrophils that may produce inflammation and tissue damage) is due to the strained conformation of the 5-membered dithiolane ring, which is lost upon reduction to DHLA. In cells, LA is reduced to dihydrolipoic acid, which is generally regarded as the more bioactive form of LA and the form responsible for most of the antioxidant effects and for lowering the redox activities of unbound iron and copper.[38] This theory has been challenged due to the high level of reactivity of the two free sulfhydryls, low intracellular concentrations of DHLA as well as the rapid methylation of one or both sulfhydryls, rapid side-chain oxidation to shorter metabolites and rapid efflux from the cell. Although both DHLA and LA have been found inside cells after administration, most intracellular DHLA probably exists as mixed disulfides with various cysteine residues from cytosolic and mitochondrial proteins.[32] Recent findings suggest therapeutic and anti-aging effects are due to modulation of signal transduction and gene transcription, which improve the antioxidant status of the cell. However, this likely occurs via pro-oxidant mechanisms, not by radical scavenging or reducing effects.[4][37][39]
All the disulfide forms of LA (R/S-LA, RLA and SLA) can be reduced to DHLA although both tissue specific and stereoselective (preference for one enantiomer over the other) reductions have been reported in model systems. At least two cytosolic enzymes, glutathione reductase (GR) and thioredoxin reductase (Trx1), and two mitochondrial enzymes, lipoamide dehydrogenase and thioredoxin reductase (Trx2), reduce LA. SLA is stereoselectively reduced by cytosolic GR whereas Trx1, Trx2 and lipoamide dehydrogenase stereoselectively reduce RLA. (R)-(+)-lipoic acid is enzymatically or chemically reduced to (R)-(-)-dihydrolipoic acid whereas (S)-(-)-lipoic acid is reduced to (S)-(+)-dihydrolipoic acid.[40][41][42][43][44][45][46] Dihydrolipoic acid (DHLA) can also form intracellularly and extracellularly via non-enzymatic, thiol-disulfide exchange reactions.[47]
RLA may function in vivo like a B-vitamin and at higher doses like plant-derived nutrients, such as curcumin, sulforaphane, resveratrol, and other nutritional substances that induce phase II detoxification enzymes, thus acting as cytoprotective agents.[39][48] This stress response indirectly improves the antioxidant capacity of the cell.[4]
The (S)-enantiomer of LA was shown to be toxic when administered to thiamine-deficient rats.[49][50]
Several studies have demonstrated that SLA either has lower activity than RLA or interferes with the specific effects of RLA by competitive inhibition.[51][52][53][54][55]

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Uses
R/S-LA and RLA are widely available as over-the-counter nutritional supplements in the United States in the form of capsules, tablets, and aqueous liquids, and have been marketed as antioxidants.[3]
Although the body can synthesize LA, it can also be absorbed from the diet. Dietary supplementation in doses from 200–600 mg is likely to provide up to 1000 times the amount available from a regular diet. Gastrointestinal absorption is variable and decreases with the use of food. It is therefore recommended that dietary LA be taken 30–60 minutes before or at least 120 minutes after a meal. Maximum blood levels of LA are achieved 30–60 minutes after dietary supplementation, and it is thought to be largely metabolized in the liver.[56]
In Germany, LA is approved as a drug for the treatment of diabetic neuropathy since 1966 and is available as a non-prescription pharmaceutical.[57]
Clinical research
According to the American Cancer Society as of 2013, “there is no reliable scientific evidence at this time that lipoic acid prevents the development or spread of cancer”.[58] As of 2015, intravenously administered ALA is unapproved anywhere in the world except Germany for diabetic neuropathy, but has been proven reasonably safe and effective in four clinical trials; however another large trial over four years found no difference from placebo.[59] As of 2012, there was no good evidence alpha lipoic acid helps people with mitochondrial disorders.[60] A 2018 review recommended ALA as an anti-obesity supplement with low dosage (< 600 mg/day) for a short period of time (<10 weeks); however, it is too expensive to be practical as a complementary therapy for obesity.[61]
SYN
WO 0210151
DE 19709069; EP 0863125; US 6013833
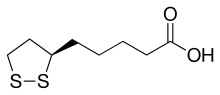
A synthetic route based on the asymmetric reduction of oxo diesters has been reported. Meldrum’s acid (LII) was acylated by methyl adipoyl chloride (LI) in the presence of pyridine to produce the intermediate (LIII) which, upon alcoholysis with isobutanol, led to oxo diester (LIV). Enantioselective reduction of (LIV) by means of baker’s yeast furnished the (S)-hydroxy diester (LV). Alternatively, the analogous oxo diester (LVI) was prepared by acylation of methyl acetoacetate with methyl adipoyl chloride (LI), followed by deacetylation in the presence of ammonium hydroxide. Then, asymmetric chemical reduction of (LVI) by hydrogenation in the presence of the chiral catalyst Ru2Cl4[(S)-BINAP]2 provided the (S)-hydroxy diester (LVII). Regioselective reduction of either diester (LV) or (LVII) by means of NaBH4 in refluxing THF furnished dihydroxy ester (XLVIII). After conversion of (XLVIII) to the dimesylate (XLIX), displacement with potassium thioacetate afforded the bis(acetylthio) derivative (LVIII), which was further hydrolyzed with KOH to provide dihydrolipoic acid (LIX). In a related procedure, dihydrolipoic acid (LIX) was prepared by reaction of dimesylate (XLIX) with sodium disulfide, followed by reductive treatment with NaBH4 and NaOH. The title cyclic disulfide was then obtained by oxidation of the dithiol (LIX) using oxygen in the presence of FeCl3.
SYN
DE 10036516; WO 0210113
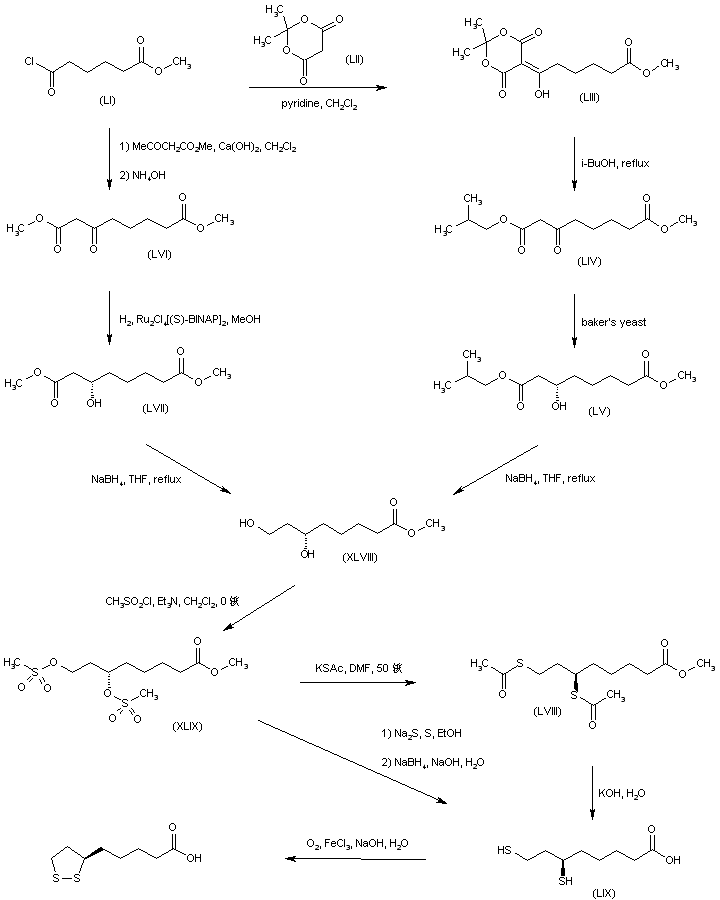
The key dihydroxy ester intermediate (XIII) was also obtained by asymmetric hydrogenation of hydroxy ketoester (XLIII) in the presence of (S)-BINAP-dichlororuthenium catalyst. The precursor hydroxy ketoester (XLIII) was prepared by two alternative procedures. In one method, the racemic dihydroxy ester (XLII) was selectively oxidized to (XLIII) by means of NaOCl. In another method, the unsaturated keto ester (XLIV) was epoxidized by means of sodium percarbonate, and the resultant epoxide (XLV) was then reduced to the hydroxy ketoester (XLIII) by catalytic hydrogenation over PtO2.
SYN
WO 0230919
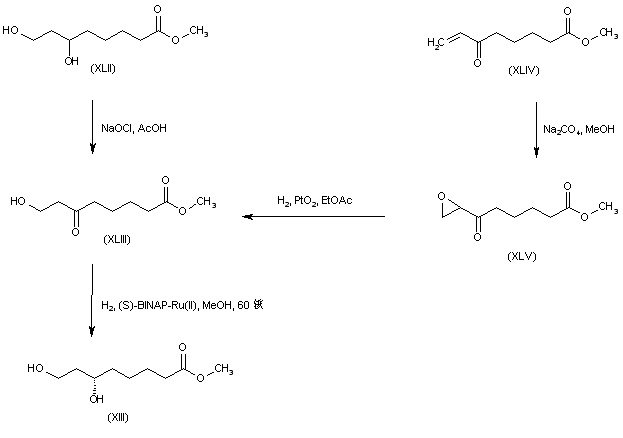
Both enantiomers of racemic 8-chloro-6-hydroxyoctanoic acid (LX) were separated employing either (+)- or (-)-alpha-methylbenzylamine. Esterification of the (R)-(-)-enantiomer with HCl-MeOH provided the chloro hydroxy ester (LXI). Further chlorination of (LXI) with SOCl2 and pyridine proceeded with inversion of configuration at C-6 to furnish the (S)-dichloro derivative (LXII). The cyclic disulfide (L) was then prepared by treatment of chloride (LXII) with sulfur and sodium sulfide in boiling EtOH. Basic hydrolysis of the methyl ester group of (LXII) then afforded (R) alpha lipoic acid. The title compound was also obtained from the (S)-(+)-acid (LXIII). Reaction of hydroxy acid (LXIII) with methanesulfonyl chloride produced the chloro mesylate (LXIV), which was then cyclized to the target disulfide in the presence of sulfur and Na2S.
SYN
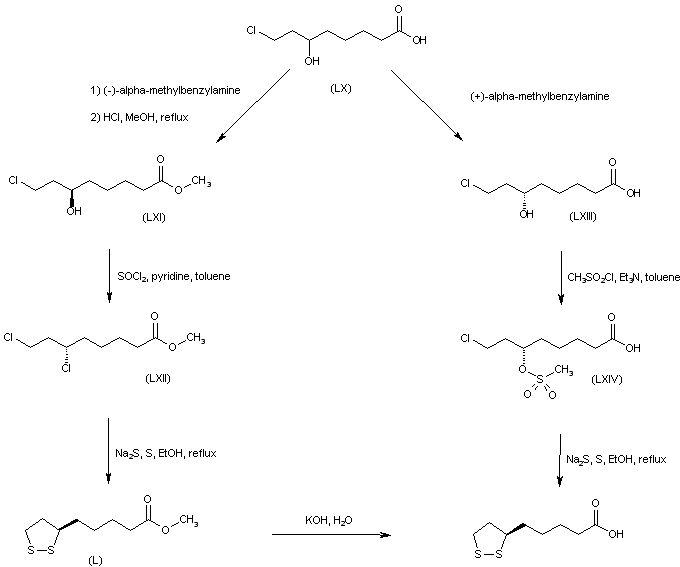
The reaction of the chiral dibenzoyloxy-dihydropyran (LXV) with H2SO4 and HgSO4 gives the unsaturated aldehyde (LXVI), which is condensed with the phosphorane (LXVII) to yield the hepatdienoic ester (LXVIII). The hydrogenation of (LXVIII) with H2 over Pd/C affords the heptanoic ester (LXIX), which is treated with Ts-Cl and pyridine to provide the tosyloxy derivative (LXX). The cyclization of (LXX) by means of K2CO3 gives the chiral epoxide (LXXI), which is condensed with vinylmagnesium bromide (LXXII) to yield 6(S)-hydroxy-8-nonenoic acid methyl ester (LXXIII). The oxidation of the terminal double bond of (LXXIII) with ozone affords the carbaldehyde (LXXIV), which is reduced with NaBH4 to provide 6(S),8-dihydroxyoctanoic acid methyl ester (XLVIII). The reaction of (XLVIII) with Ms-Cl and pyridine gives the dimesylate (XLIX), which is treated with Na2S2 to yield the lipoic acid methyl ester (L), which is hydrolyzed to the target acid with KOH in H2O.
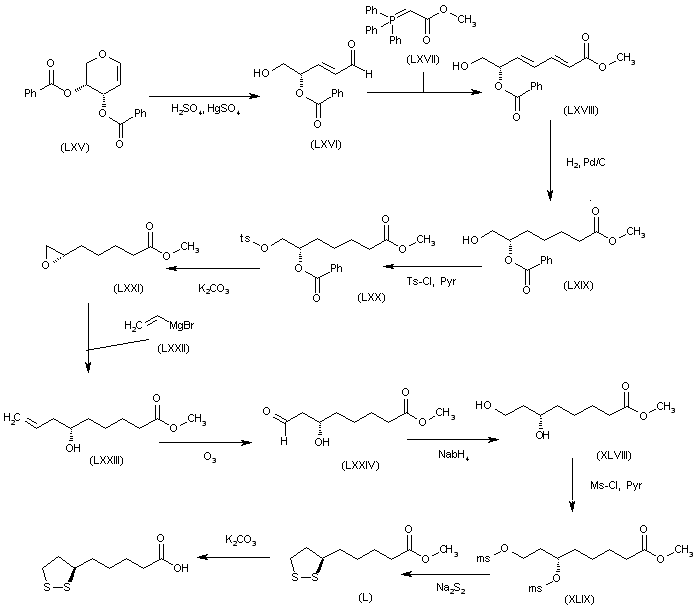
SYN
DE 3629116; EP 0261336
Alkylation of the lithio-dianion of propargyl alcohol (XIII) with 6-bromo-1-hexene (XIV), followed by in situ reduction of the resultant disubstituted acetylene with lithium metal gave the allylic alcohol (XV). Asymmetric Sharpless epoxidation of (XV) using tert-butyl hydroperoxide in the presence of L-(+)-diisopropyl tartrate afforded the (S,S)-epoxy alcohol (XVI). This was reduced to the chiral diol (XVII) employing Red-Al?in THF. After formation of the bis-mesylate (XVIII), oxidative cleavage of the terminal double bond by means of NaIO4 in the presence of ruthenium catalyst furnished the carboxylic acid (XIX). The mesylate groups were finally displaced by sodium disulfide to produce the desired cyclic disulfide compound.
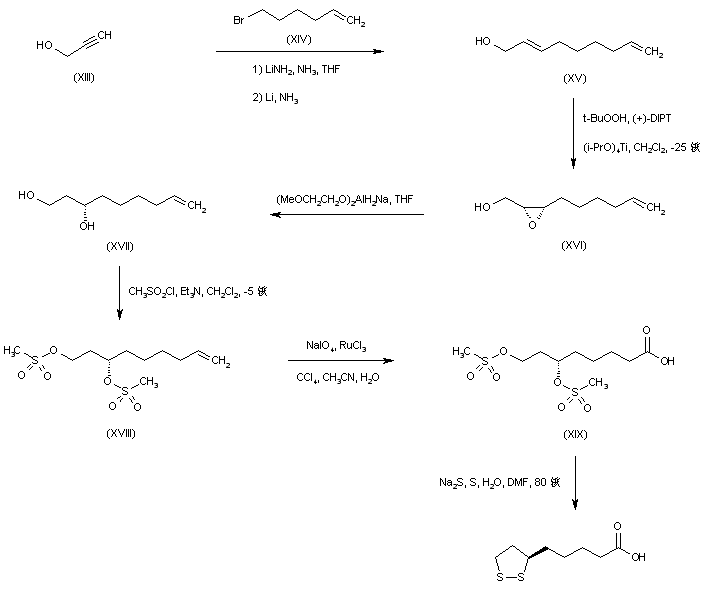
SYN
Both enantiomers of racemic 8-chloro-6-hydroxyoctanoic acid (LX) were separated employing either (+)- or (-)-alpha-methylbenzylamine. Esterification of the (R)-(-)-enantiomer with HCl-MeOH provided the chloro hydroxy ester (LXI). Further chlorination of (LXI) with SOCl2 and pyridine proceeded with inversion of configuration at C-6 to furnish the (S)-dichloro derivative (LXII). The cyclic disulfide (L) was then prepared by treatment of chloride (LXII) with sulfur and sodium sulfide in boiling EtOH. Basic hydrolysis of the methyl ester group of (LXII) then afforded (R) alpha lipoic acid. The title compound was also obtained from the (S)-(+)-acid (LXIII). Reaction of hydroxy acid (LXIII) with methanesulfonyl chloride produced the chloro mesylate (LXIV), which was then cyclized to the target disulfide in the presence of sulfur and Na2S.
| DE 19533881; EP 0763533; US 5731448 |
SYN
WO 9638437
A different strategy was based on the enantioselective oxidation of a cyclohexanone derivative by enzymic Baeyer-Villiger reaction. Keto ester (XXXVIII) was protected as the ethylene ketal (XXXIX) and subsequently reduced to alcohol (XL) using LiAlH4. Acetylation of alcohol (XL) to acetate (XLI), followed by acidic ketal hydrolysis afforded cyclohexanone (XLII) (9,10). The racemic ketone (XLII) was then subjected to oxidative cleavage by monooxigenase 2 obtained from Pseudomonas putida to furnish the (R)-lactone (XLIV) along with unreacted (S)-cyclohexanone (XLIII) (9-11). The use of cyclohexanone monooxigenase from Acinetobacter NCIMB 9871 has also been reported for this reaction (12). Methanolysis of lactone (XLIV) in the presence of NaOMe gave rise to the (R)-dihydroxy ester (XLV). Inversion of the configuration of (XLV) was accomplished by Mitsunobu coupling with p-nitrobenzoic acid (XLVI) to produce the (S)-p-nitrobenzoate ester (XLVII). Smooth hydrolysis of ester (XLVII) provided methyl (S)-6,8-dihydroxyoctanoate (XLVIII), which was processed through intermediates (XLIX) and (L), as for the isopropyl (X) (Scheme 29605101a) and ethyl (XXIX) (Scheme 29605103a) homologues, to afford the title compound.
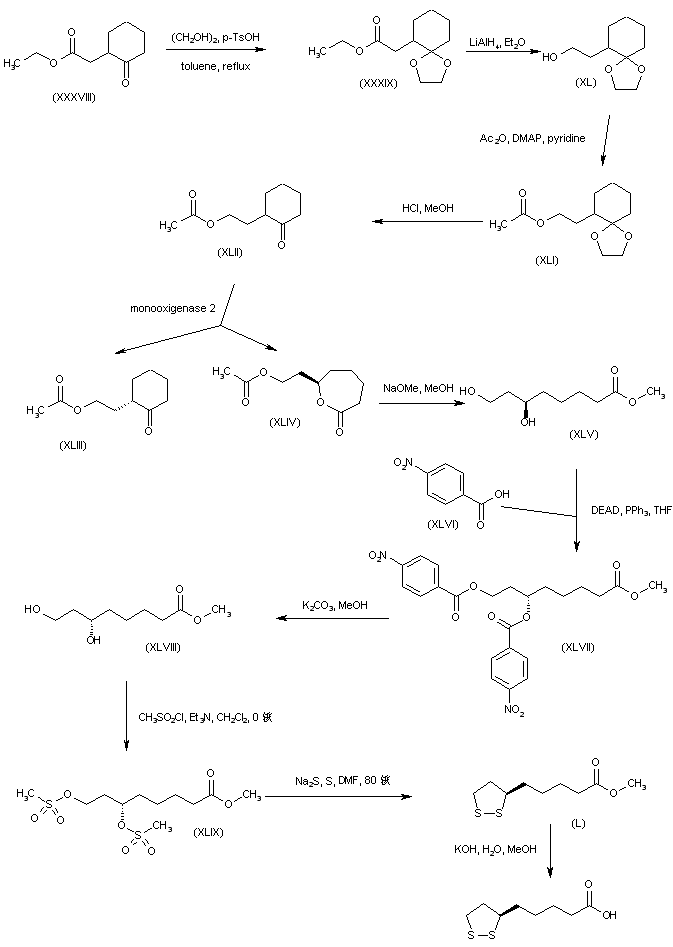
SYN
| Tetrahedron Lett 2001,42(29),4891 |
The olefinic diester (XXXVIII) was subjected to OsO4-catalyzed asymmetric dihydroxylation using hydroquinidine 1,4-phthalazinediyl diether [(DHQD)2-PHAL] as chiral ligand to afford diol (XXXIX). This was converted to the cyclic sulfate (XL) by treatment with SOCl2, followed by RuCl3-catalyzed NaIO4 oxidation of the intermediate sulfite. Regioselective reduction of sulfate (XL) at the alpha position with NaBH4 in DMA led to the (3S)-alcohol (XLI). Further selective reduction of the ethyl ester group of (XLI) was achieved by treatment with NaBH4-Et3N in MeOH-DMF, yielding the target intermediate dihydroxy ester (XIII).
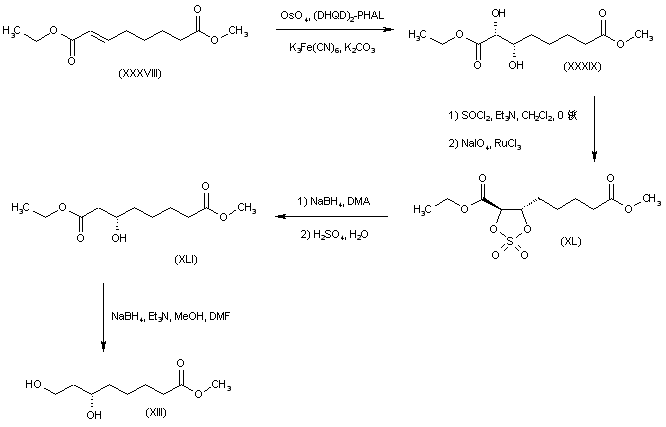
SYN
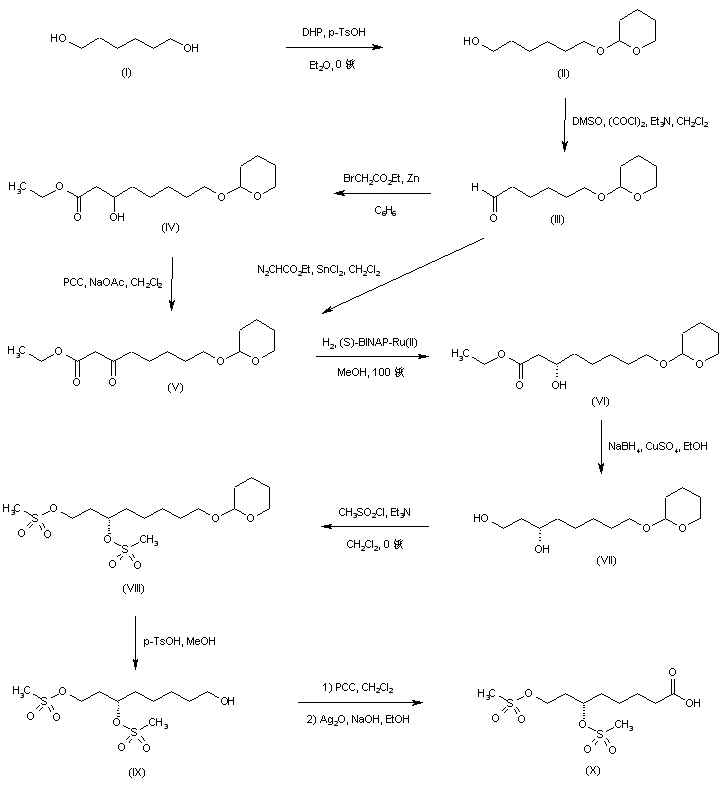
1,6-Hexanediol (I) was protected as the mono-tetrahydropyranyl ether (II), and the free hydroxyl group was subsequently oxidized to aldehyde (III) under Swern conditions. Reformatskii reaction of aldehyde (III) with the organozinc reagent generated from ethyl bromoacetate yielded the racemic hydroxy ester (IV). The requisite (S)-enantiomer (VI) was obtained via oxidation of (IV) to oxo ester (V) using pyridinium chlorochromate, and then asymmetric hydrogenation in the presence of (S)-(-)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl dichlororuthenium complex. Oxo ester (V) was also prepared by SnCl2-catalyzed insertion of ethyl diazoacetate into aldehyde (III). The chiral hydroxy ester (VI) was then reduced to diol (VII) by means of NaBH4-CuSO4. After conversion of (VII) to the corresponding dimesylate (VIII), removal of the tetrahydropyranyl protecting group under acidic conditions gave alcohol (IX). This was sequentially oxidized with PCC to aldehyde, and then with Ag2O to furnish the target dimesylate acid intermediate (X).
SYN
Tetrahedron Asymmetry 2000,11(4),879
The intermediate 6(S)-hydroxy-8-nonenoic acid methyl ester (III) has been obtained by enantioselective allylation of 6-oxohexanoic acid methyl ester (I) with allyltributylstannane (II) catalyzed by the chiral catalyst (R)-BINOL/Ti(O-iPr)4 in refluxing dichloromethane (other BINOL/metal catalysts have also been studied).
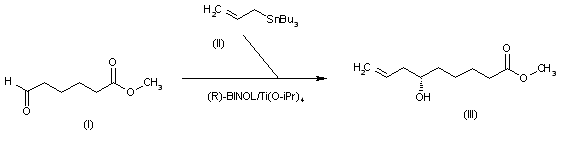
SYN
Tetrahedron Lett 1985,26(21),2535
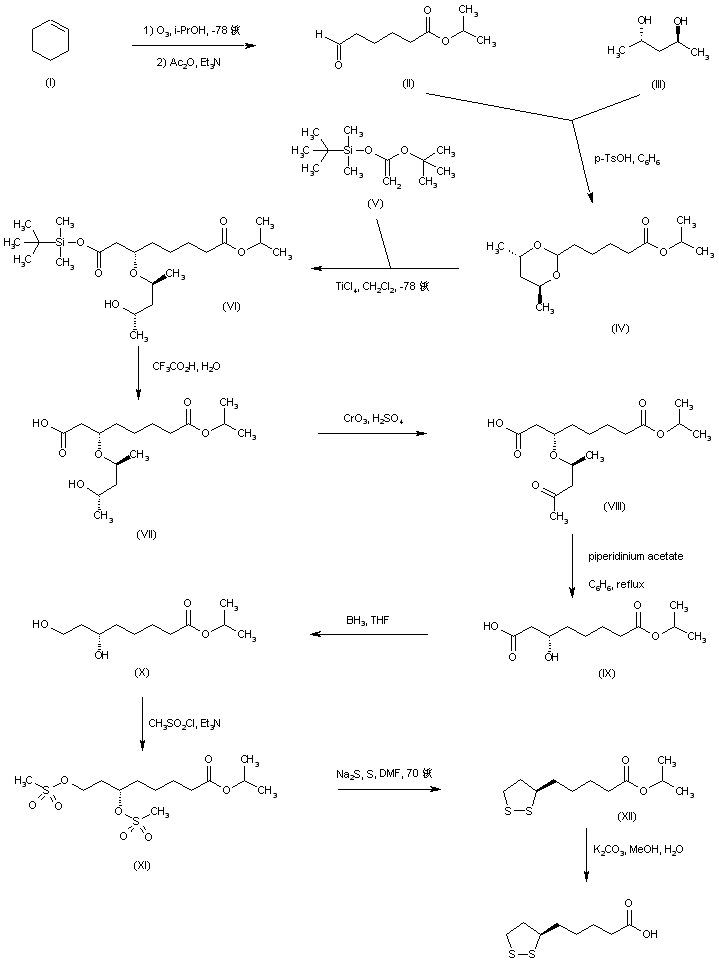
Aldehyde (II), prepared by ozonolysis of cyclohexene (I), was ketalized with (S,S)-2,4-pentanediol (III) to afford dioxane (IV). Titanium chloride-mediated coupling of acetal (IV) with the ketene acetal (V) afforded diastereoselectively adduct (VI), which was subsequently hydrolyzed to carboxylic acid (VII) by means of trifluoroacetic acid. Removal of the pentanediol moiety to furnish the (R)-alcohol (IX) was accomplished via Jones oxidation of the secondary alcohol (VII) to ketone (VIII), followed by beta-elimination in the presence of piperidinium acetate. Reduction of the free carboxyl group by borane-tetrahydrofuran complex gave diol (X), which was further converted to dimesylate (XI). Disulfide displacement of the mesylate groups provided (+)-lipoic acid isopropyl ester (XII), which was finally hydrolyzed to the title acid using K2CO3 in MeOH/H2O.
SYN
Tetrahedron Lett 1987,28(44),5313
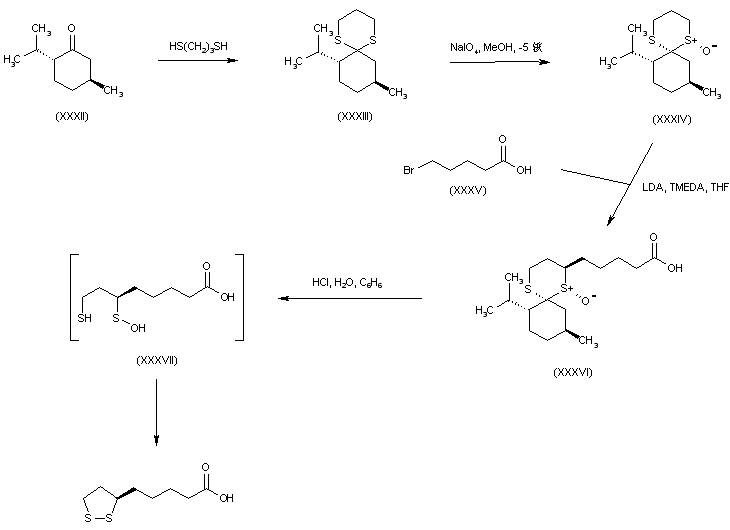
A short synthetic strategy utilized the cyclic thioketal (XXXIII), derived from d-menthone (XXXII) and 1,3-propanedithiol, as the chiral template. Stereospecific oxidation of dithiane (XXXIII) employing NaIO4 produced sulfoxide (XXXIV). The carbanion generated from sulfoxide (XXXIV) was stereoselectively alkylated by 5-bromopentanoic acid (XXXV) in the presence of TMEDA to furnish the trans alkylated compound (XXXVI). Finally, acidic hydrolysis of (XXXVI) formed the intermediate mercapto sulfinic acid (XXXVII) which spontaneously cyclized to the desired dithiolane derivative.
SYN
Tetrahedron Lett 1987,28(19),2183
Diisopropylidene mannitol (I) was first converted into the dibutyltin derivative (II), which was subsequently mono-benzylated to (III). Acetylation of (III) with acetic anhydride in pyridine gave (IV). After acidic hydrolysis of the isopropylidene ketals of (IV), the resultant tetraol (V) was converted into tetramesylate (VI). Reductive elimination in (VI) with Zn and NaI produced diene (VII). The acetate group of (VII) was then hydrolyzed to (VIII) using NaOMe. Intermediate (VIII) was reacted with triethyl orthoacetate in the presence of propionic acid to generate the allyl vinyl ether (IX), which underwent a Claisen rearrangement to the diene-ester (X). Selective hydroboration-oxidation of the terminal double bond of (X) yielded the primary alcohol (XI). Subsequent benzyl group hydrogenolysis in (XI) furnished the target intermediate diol (XII).
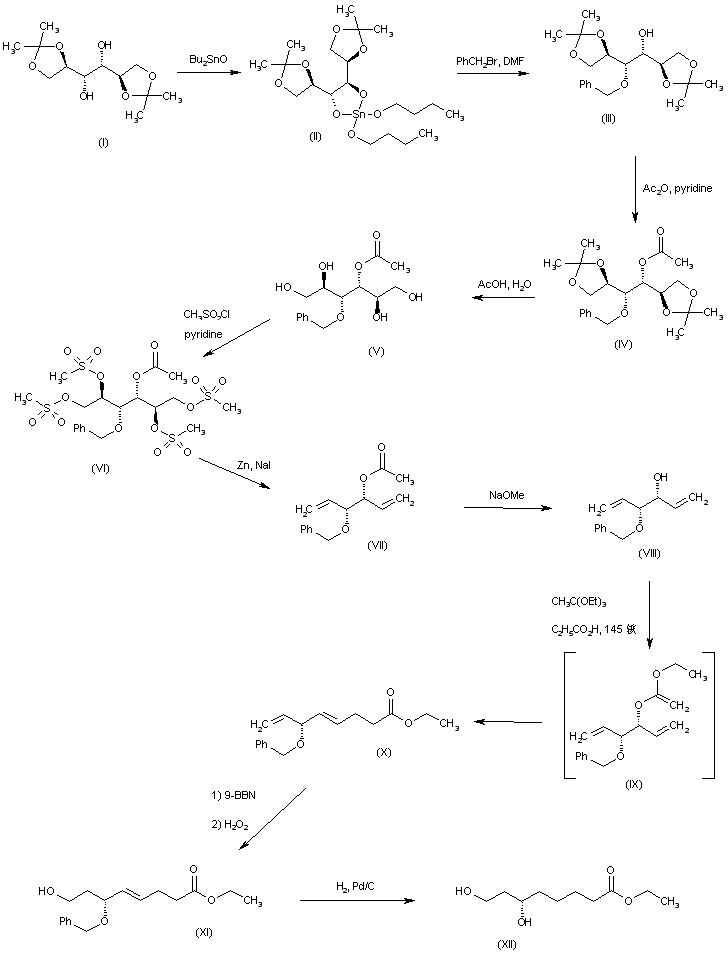
SYN
Esterification of diisopropylidene mannitol (I) with benzoyl chloride in pyridine afforded dibenzoate (II). Hydrolysis of the isopropylidene ketals of (II) with aqueous HOAc gave tetraol (III), which was further converted to tetramesylate (IV) on treatment with methanesulfonyl chloride and pyridine. Reductive elimination of the mesylate groups of (IV) using Zn dust and NaI yielded diene (V). The benzoate esters of (V) were then removed by treatment with sodium methoxide. The resultant divinylglycol (VI) was reacted with dibutyltin oxide to produce the tin derivative (VII), which was converted to the target intermediate, themono-benzyl ether (VIII), by treatment with benzyl bromide in hot DMF.
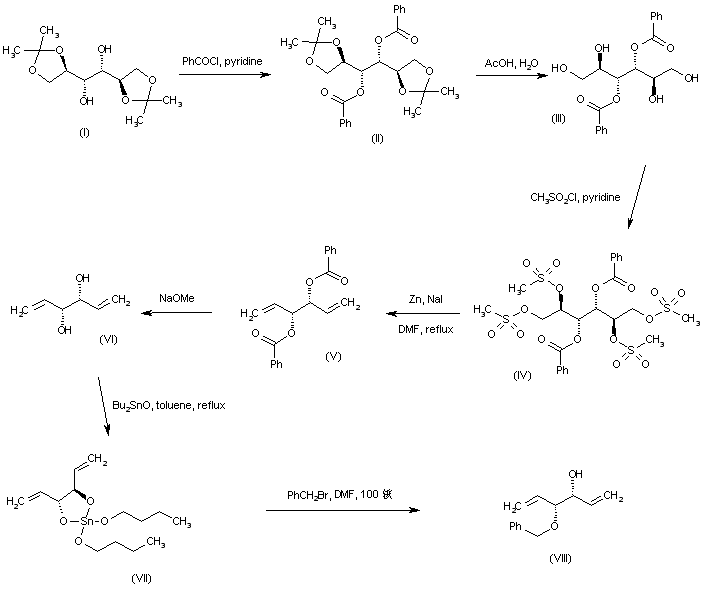
SYN
Tetrahedron Lett 1989,30(42),5705
Alkylation of the dianion of octyl acetoacetate (XIII) with 4-iodobutyronitrile (XIV) provided the cyano keto ester (XV). Enantiospecific reduction of (XV) utilizing baker’s yeast gave rise to the desired (S)-hydroxy ester (XVI) in high enantiomeric excess. Subsequent ester group reduction in (XVI) by means of LiBH4 provided diol (XVII). The target dihydroxy ester (XII) was then obtained by alcoholysis of nitrile (XVII) under acidic conditions.
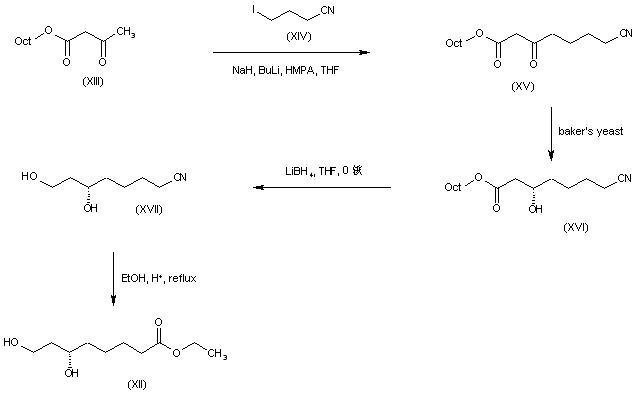
SYN
J Chem Soc Chem Commun 1995,(15),1563
A different strategy was based on the enantioselective oxidation of a cyclohexanone derivative by enzymic Baeyer-Villiger reaction. Keto ester (XXXVIII) was protected as the ethylene ketal (XXXIX) and subsequently reduced to alcohol (XL) using LiAlH4. Acetylation of alcohol (XL) to acetate (XLI), followed by acidic ketal hydrolysis afforded cyclohexanone (XLII) (9,10). The racemic ketone (XLII) was then subjected to oxidative cleavage by monooxigenase 2 obtained from Pseudomonas putida to furnish the (R)-lactone (XLIV) along with unreacted (S)-cyclohexanone (XLIII) (9-11). The use of cyclohexanone monooxigenase from Acinetobacter NCIMB 9871 has also been reported for this reaction (12). Methanolysis of lactone (XLIV) in the presence of NaOMe gave rise to the (R)-dihydroxy ester (XLV). Inversion of the configuration of (XLV) was accomplished by Mitsunobu coupling with p-nitrobenzoic acid (XLVI) to produce the (S)-p-nitrobenzoate ester (XLVII). Smooth hydrolysis of ester (XLVII) provided methyl (S)-6,8-dihydroxyoctanoate (XLVIII), which was processed through intermediates (XLIX) and (L), as for the isopropyl (X) (Scheme 29605101a) and ethyl (XXIX) (Scheme 29605103a) homologues, to afford the title compound.
SYN
Synthesis (Stuttgart) 1996,(5),594
Racemic tetrahydro-2-furylmethanol (I) was converted to tosylate (II), which was further displaced by KCN to yield nitrile (III). Basic hydrolysis of nitrile (III), followed by Fischer esterification of the resultant carboxylic acid (IV) provided ethyl ester (V). Enzymatic resolution of racemic ester (V) by means of the lipase from Candida cylindracea generated a mixture of the (R)-acid (VI) and the unreacted (S)-ester (VII), which were separated by column chromatography. The desired (S) ester (VII) was then reduced to alcohol (VIII) with LiAlH4 in cold Et2O. Regioselective opening of the cyclic ether (VIII) with iodotrimethylsilane in acetone furnished the acetonide of 6-iodo-1,3-hexanediol (IX). Alkylation of benzyl methyl malonate (X) with iodide (IX) provided malonate (XI). Hydrogenolysis of the benzyl ester group of (XI), followed by thermal decarboxylation led to ester (XII). The target dihydroxy ester precursor (XIII) was then obtained by acid-catalyzed hydrolysis of the acetonide function.
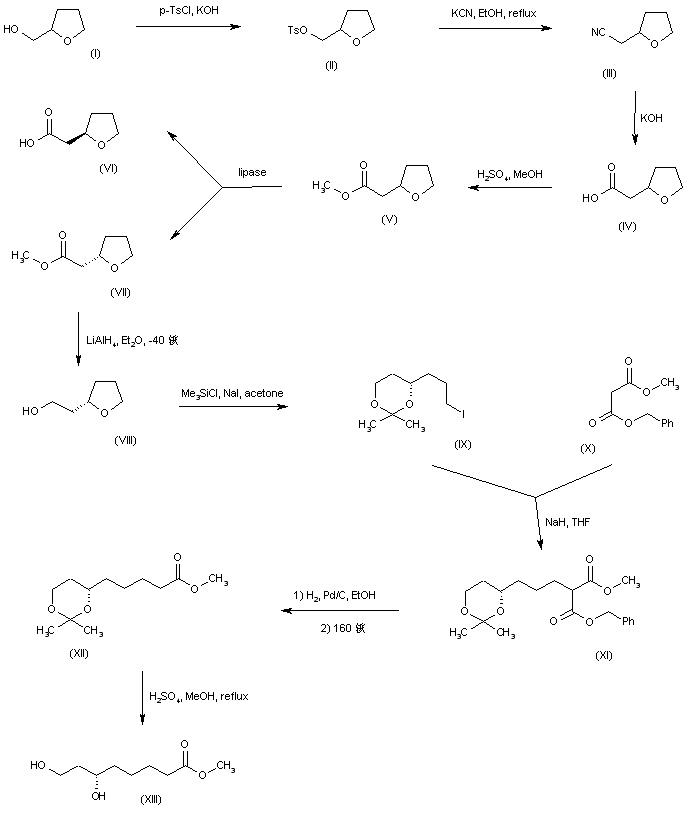
SYN
Synthesis (Stuttgart) 1996,(11),1289
Addition of vinylmagnesium bromide to 2-nitrocyclohexanone (XIV) afforded the nitro alcohol (XV). Ring cleavage of (XVI) in the presence of anhydrous CuSO4 absorbed on silica gel gave the nitro ketone (XVI). Nitro group hydrolysis in (XVI) by successive treatment with NaOMe and H2SO4 in MeOH furnished oxo ester (XVII) as the main product. This was enantiospecifically reduced with baker’s yeast to yield the (S)-alcohol (XVIII). Selective methyl ether cleavage with tetrabutylammonium iodide and BF3 provided the dihydroxy ester precursor (XIII).
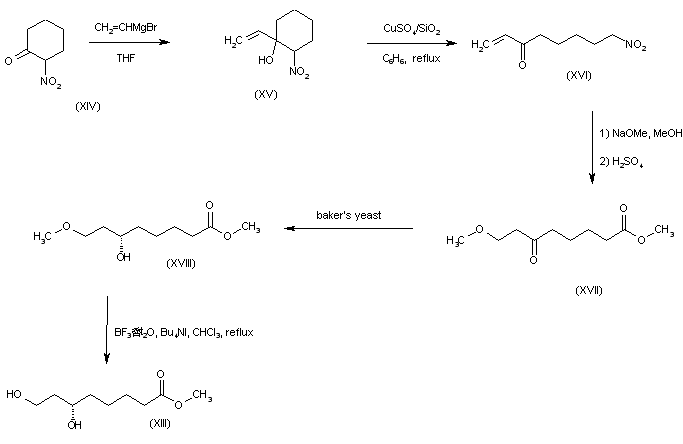
SYN
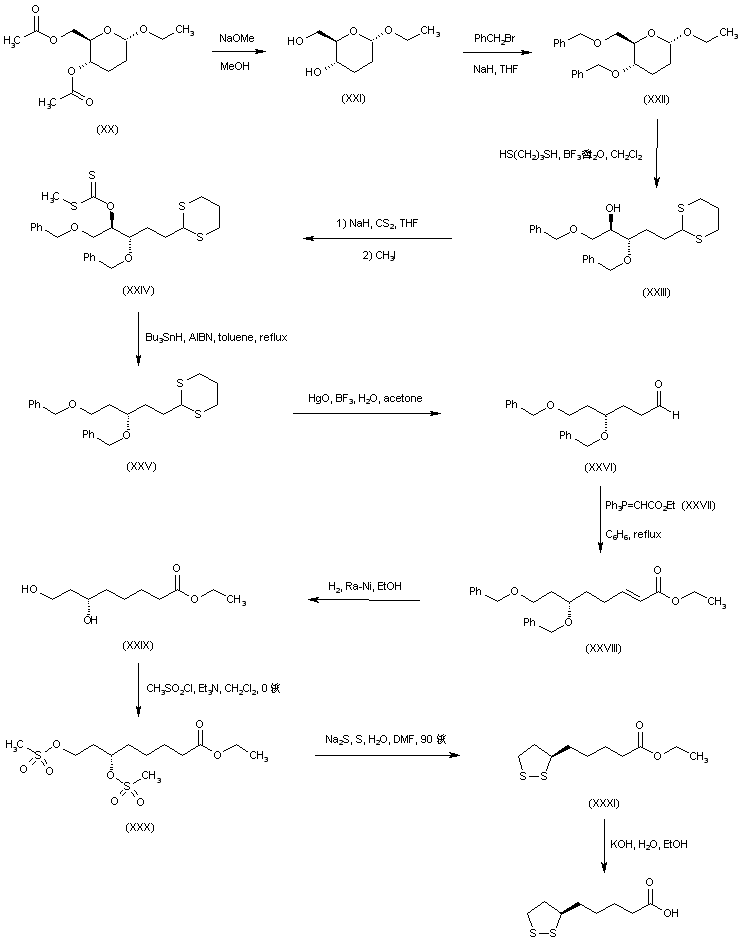
An alternative route to (+)-lipoic acid used ethyl 4,6-di-O-acetyl-2,3-dideoxy-alpha-D-erythro-hexopyranoside (XX), prepared from triacetyl-D-glucal, as the chiral starting point. Deacetylation of (XX) with sodium methoxide under Zemplen conditions gave diol (XXI) which, after conventional benzylation, led to the 4,6-di-O-benzyl derivative (XXII). Ring opening of the cyclic acetal (XXII) with propanediol in the presence of boron trifluoride afforded the dithiane derivative (XXIII). The free hydroxyl group of (XXIII) was converted into xanthate (XXIV) by reaction with NaH and CS2, followed by methyl iodide. Reductive cleavage of the xanthate group by means of Bu3SnH and AIBN provided (XXV). Hydrolysis of the thioacetal function with HgO and BF3 provided aldehyde (XXVI). Chain homologation was performed by Wittig reaction of aldehyde (XXVI) with phosphorane (XXVII) to afford the unsaturated ester (XXVIII). Simultaneous double bond hydrogenation and benzyl ether cleavage in the presence of Raney nickel led to dihydroxy ester (XXIX). This was converted to the corresponding dimesylate (XXX), which was further cyclized to disulfide (XXXI) using the in situ generated sodium disulfide as in the precedent Schemes. Finally, basic hydrolysis of the ethyl ester (XXXI) yielded the title carboxylic acid.
| Carbohydr Res 1986,148(1),51 |
SYN
Diisopropylidene mannitol (I) was first converted into the dibutyltin derivative (II), which was subsequently mono-benzylated to (III). Acetylation of (III) with acetic anhydride in pyridine gave (IV). After acidic hydrolysis of the isopropylidene ketals of (IV), the resultant tetraol (V) was converted into tetramesylate (VI). Reductive elimination in (VI) with Zn and NaI produced diene (VII). The acetate group of (VII) was then hydrolyzed to (VIII) using NaOMe. Intermediate (VIII) was reacted with triethyl orthoacetate in the presence of propionic acid to generate the allyl vinyl ether (IX), which underwent a Claisen rearrangement to the diene-ester (X). Selective hydroboration-oxidation of the terminal double bond of (X) yielded the primary alcohol (XI). Subsequent benzyl group hydrogenolysis in (XI) furnished the target intermediate diol (XII).
| J Carbohydr Chem 1990,9(2-3),307 |
SYN
J Chem Soc Chem Commun 1986,(18),1408
SYN
https://www.sciencedirect.com/science/article/abs/pii/S1381117713003342

References
- ^ “Lipoic Acid”. Pubmed. NCBI. Retrieved October 18, 2018.
- ^ Teichert, J; Hermann, R; Ruus, P; Preiss, R (November 2003). “Plasma kinetics, metabolism, and urinary excretion of alpha-lipoic acid following oral administration in healthy volunteers”. The Journal of Clinical Pharmacology. 43 (11): 1257–67. doi:10.1177/0091270003258654. PMID 14551180. S2CID 30589232.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y “Lipoic acid”. Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis. 1 January 2019. Retrieved 5 November 2019.
- ^ Jump up to:a b c d e Shay, KP; Moreau, RF; Smith, EJ; Hagen, TM (June 2008). “Is alpha-lipoic acid a scavenger of reactive oxygen species in vivo? Evidence for its initiation of stress signaling pathways that promote endogenous antioxidant capacity”. IUBMB Life. 60 (6): 362–7. doi:10.1002/iub.40. PMID 18409172. S2CID 33008376.
- ^ Reljanovic, M; Reichel, G; Rett, K; Lobisch, M; et al. (September 1999). “Treatment of diabetic polyneuropathy with the antioxidant thioctic acid (alpha-lipoic acid): A two year multicenter randomized double-blind placebo-controlled trial (ALADIN II). Alpha Lipoic Acid in Diabetic Neuropathy”. Free Radical Research. 31 (3): 171–9. doi:10.1080/10715769900300721. PMID 10499773.
- ^ USP32-NF27. p. 1042.
- ^ “Pharmacopeial Forum”. 34 (5): 1209.
- ^ Jiang, Y; Cronan, JE (2005). “Expression cloning and demonstration of Enterococcus faecalis lipoamidase (pyruvate dehydrogenase inactivase) as a Ser-Ser-Lys triad amidohydrolase”. Journal of Biological Chemistry. 280 (3): 2244–56. doi:10.1074/jbc.M408612200. PMID 15528186.
- ^ Cronan, JE; Zhao, X; Jiang, Y (2005). Poole, RK (ed.). Function, attachment and synthesis of lipoic acid in Escherichia coli. Advances in Microbial Physiology. 50. pp. 103–46. doi:10.1016/S0065-2911(05)50003-1. ISBN 9780120277506. PMID 16221579.
- ^ Zempleni, J.; Trusty, T. A.; Mock, D. M. (1997). “Lipoic acid reduces the activities of biotin-dependent carboxylases in rat liver”. The Journal of Nutrition. 127 (9): 1776–81. doi:10.1093/jn/127.9.1776. PMID 9278559.
- ^ Chirapu, S. R.; Rotter, C. J.; Miller, E. L.; Varma, M. V.; Dow, R. L.; Finn, M. G. (2013). “High specificity in response of the sodium-dependent multivitamin transporter to derivatives of pantothenic acid”. Current Topics in Medicinal Chemistry. 13 (7): 837–42. doi:10.2174/1568026611313070006. PMID 23578027.
- ^ Machado, RS; Clark, DP; Guest, JR (1992). “Construction and properties of pyruvate dehydrogenase complexes with up to nine lipoyl domains per lipoate acetyltransferase chain”. FEMS Microbiology Letters. 79 (1–3): 243–8. doi:10.1111/j.1574-6968.1992.tb14047.x. PMID 1478460.
- ^ Douce, R; Bourguignon, J; Neuburger, M; Rebeille, F (2001). “The glycine decarboxylase system: A fascinating complex”. Trends in Plant Science. 6 (4): 167–76. doi:10.1016/S1360-1385(01)01892-1. PMID 11286922.
- ^ Durrani, AI; Schwartz, H; Nagl, M; Sontag, G (October 2010). “Determination of free [alpha]-lipoic acid in foodstuffs by HPLC coupled with CEAD and ESI-MS”. Food Chemistry. 120 (4): 38329–36. doi:10.1016/j.foodchem.2009.11.045.
- ^ Reed, LJ (October 2001). “A trail of research from lipoic acid to alpha-keto acid dehydrogenase complexes”. Journal of Biological Chemistry. 276 (42): 38329–36. doi:10.1074/jbc.R100026200. PMID 11477096.
- ^ Hermann, R; Niebch, G; Borbe, HO; Fieger, H; et al. (1996). “Enantioselective pharmacokinetics and bioavailability of different racemic formulations in healthy volunteers”. European Journal of Pharmaceutical Sciences. 4 (3): 167–74. doi:10.1016/0928-0987(95)00045-3.
- ^ Teichert, J; Preiss, R (1997). High-performance Liquid Chromatography Methods for Determination of Lipoic and Dihydrolipoic Acid in Human Plasma. Methods in Enzymology. 279. pp. 159–66. doi:10.1016/S0076-6879(97)79019-0. ISBN 9780121821807. PMID 9211267.
- ^ Teichert, J; Preiss, R (October 1995). “Determination of lipoic acid in human plasma by high-performance liquid chromatography with electrochemical detection”. Journal of Chromatography B. 672 (2): 277–81. doi:10.1016/0378-4347(95)00225-8. PMID 8581134.
- ^ Teichert, J; Preiss, R (November 1992). “HPLC-methods for determination of lipoic acid and its reduced form in human plasma”. International Journal of Clinical Pharmacology, Therapy, and Toxicology. 30 (11): 511–2. PMID 1490813.
- ^ Biewenga, GP; Haenen, GR; Bast, A (September 1997). “The pharmacology of the antioxidant lipoic acid”. General Pharmacology. 29 (3): 315–31. doi:10.1016/S0306-3623(96)00474-0. PMID 9378235.
- ^ Jump up to:a b Schupke, H; Hempel, R; Peter, G; Hermann, R; et al. (June 2001). “New metabolic pathways of alpha-lipoic acid”. Drug Metabolism and Disposition. 29 (6): 855–62. PMID 11353754.
- ^ Teichert, J; Hermann, R; Ruus, P; Preiss, R (November 2003). “Plasma kinetics, metabolism, and urinary excretion of alpha-lipoic acid following oral administration in healthy volunteers”. Journal of Clinical Pharmacology. 43 (11): 1257–67. doi:10.1177/0091270003258654. PMID 14551180. S2CID 30589232.
- ^ Hornberger, CS; Heitmiller, RF; Gunsalus, IC; Schnakenberg, GHF; et al. (1953). “Synthesis of DL—lipoic acid”. Journal of the American Chemical Society. 75 (6): 1273–7. doi:10.1021/ja01102a003.
- ^ Hornberger, CS; Heitmiller, RF; Gunsalus, IC; Schnakenberg, GHF; et al. (1952). “Synthetic preparation of lipoic acid”. Journal of the American Chemical Society. 74 (9): 2382. doi:10.1021/ja01129a511.
- ^ Jump up to:a b Kleeman, A; Borbe, HO; Ulrich, H (1991). “Thioctic Acid-Lipoic Acid”. In Borbe, HO; Ulrich, H (eds.). Thioctsäure: Neue Biochemische, Pharmakologische und Klinische Erkenntnisse zur Thioctsäure [Thioctic Acid. New Biochemistry, Pharmacology and Findings from Clinical Practice with Thioctic Acid]. Symposium at Wiesbaden, DE, 16–18 February 1989. Frankfurt, DE: Verlag. pp. 11–26. ISBN 9783891191255.
- ^ Fontanella, L (1955). “Preparation of optical antipodes of alpha-lipoic acid”. Il Farmaco; Edizione Scientifica. 10 (12): 1043–5. PMID 13294188.
- ^ Walton, E; Wagner, AF; Bachelor, FW; Peterson, LH; et al. (1955). “Synthesis of (+)-lipoic acid and its optical antipode”. Journal of the American Chemical Society. 77 (19): 5144–9. doi:10.1021/ja01624a057.
- ^ Acker, DS; Wayne, WJ (1957). “Optically active and radioactive α-lipoic acids”. Journal of the American Chemical Society. 79 (24): 6483–6487. doi:10.1021/ja01581a033.
- ^ Deguchi, Y; Miura, K (June 1964). “Studies on the synthesis of thioctic acid and its related compounds. XIV. Synthesis of (+)-thioctamide”. Yakugaku Zasshi. 84 (6): 562–3. doi:10.1248/yakushi1947.84.6_562. PMID 14207116.
- ^ Lang, G (1992). In Vitro Metabolism of a-Lipoic Acid Especially Taking Enantioselective Bio-transformation into Account (Ph.D. thesis). Münster, DE: University of Münster.
- ^ US patent 5281722, Blaschke, G; Scheidmantel, U & Bethge, H et al., “Preparation and use of salts of the pure enantiomers of alpha-lipoic acid”, issued 1994-01-25, assigned to DeGussa.
- ^ Jump up to:a b Carlson, DA; Young, KL; Fischer, SJ; Ulrich, H. “Ch. 10: An Evaluation of the Stability and Pharmacokinetics of R-lipoic Acid and R-Dihydrolipoic Acid Dosage Forms in Plasma from Healthy Human Subjects”. Lipoic Acid: Energy Production, Antioxidant Activity and Health Effects. pp. 235–70. In Packer & Patel 2008.
- ^ Packer, L; Kraemer, K; Rimbach, G (October 2001). “Molecular aspects of lipoic acid in the prevention of diabetes complications”. Nutrition. 17 (10): 888–95. doi:10.1016/S0899-9007(01)00658-X. PMID 11684397.
- ^ Jump up to:a b c Carlson, DA; Smith, AR; Fischer, SJ; Young, KL; et al. (December 2007). “The plasma pharmacokinetics of R-(+)-lipoic acid administered as sodium R-(+)-lipoate to healthy human subjects” (PDF). Alternative Medicine Review. 12 (4): 343–51. PMID 18069903.
- ^ Hill, AS; Werner, JA; Rogers, QR; O’Neill, SL; et al. (April 2004). “Lipoic acid is 10 times more toxic in cats than reported in humans, dogs or rats”. Journal of Animal Physiology and Animal Nutrition. 88 (3–4): 150–6. doi:10.1111/j.1439-0396.2003.00472.x. PMID 15059240.
- ^ Packer, L; Witt, EH; Tritschler, HJ (August 1995). “Alpha-lipoic acid as a biological antioxidant”. Free Radical Biology and Medicine. 19 (2): 227–50. doi:10.1016/0891-5849(95)00017-R. PMID 7649494.
- ^ Jump up to:a b Shay, KP; Moreau, RF; Smith, EJ; Smith, AR; et al. (October 2009). “Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential”. Biochimica et Biophysica Acta (BBA) – General Subjects. 1790 (10): 1149–60. doi:10.1016/j.bbagen.2009.07.026. PMC 2756298. PMID 19664690.
- ^ Haenen, GRMM; Bast, A (1991). “Scavenging of hypochlorous acid by lipoic acid”. Biochemical Pharmacology. 42 (11): 2244–6. doi:10.1016/0006-2952(91)90363-A. PMID 1659823.
- ^ Jump up to:a b Shay, KP; Shenvi, S; Hagen, TM. “Ch. 14 Lipoic Acid as an Inducer of Phase II Detoxification Enzymes Through Activation of Nr-f2 Dependent Gene Expression”. Lipoic Acid: Energy Production, Antioxidant Activity and Health Effects. pp. 349–71. In Packer & Patel 2008.
- ^ Arnér, ES; Nordberg, J; Holmgren, A (August 1996). “Efficient reduction of lipoamide and lipoic acid by mammalian thioredoxin reductase”. Biochemical and Biophysical Research Communications. 225 (1): 268–74. doi:10.1006/bbrc.1996.1165. PMID 8769129.
- ^ Biaglow, JE; Ayene, IS; Koch, CJ; Donahue, J; et al. (April 2003). “Radiation response of cells during altered protein thiol redox”. Radiation Research. 159 (4): 484–94. Bibcode:2003RadR..159..484B. doi:10.1667/0033-7587(2003)159[0484:RROCDA]2.0.CO;2. PMID 12643793.
- ^ Haramaki, N; Han, D; Handelman, GJ; Tritschler, HJ; et al. (1997). “Cytosolic and mitochondrial systems for NADH- and NADPH-dependent reduction of alpha-lipoic acid”. Free Radical Biology and Medicine. 22 (3): 535–42. doi:10.1016/S0891-5849(96)00400-5. PMID 8981046.
- ^ Constantinescu, A; Pick, U; Handelman, GJ; Haramaki, N; et al. (July 1995). “Reduction and transport of lipoic acid by human erythrocytes”. Biochemical Pharmacology. 50 (2): 253–61. doi:10.1016/0006-2952(95)00084-D. PMID 7632170.
- ^ May, JM; Qu, ZC; Nelson, DJ (June 2006). “Cellular disulfide-reducing capacity: An integrated measure of cell redox capacity”. Biochemical and Biophysical Research Communications. 344 (4): 1352–9. doi:10.1016/j.bbrc.2006.04.065. PMID 16650819.
- ^ Jones, W; Li, X; Qu, ZC; Perriott, L; et al. (July 2002). “Uptake, recycling, and antioxidant actions of alpha-lipoic acid in endothelial cells”. Free Radical Biology and Medicine. 33 (1): 83–93. doi:10.1016/S0891-5849(02)00862-6. PMID 12086686.
- ^ Schempp, H; Ulrich, H; Elstner, EF (1994). “Stereospecific reduction of R(+)-thioctic acid by porcine heart lipoamide dehydrogenase/diaphorase”. Zeitschrift für Naturforschung C. 49 (9–10): 691–2. doi:10.1515/znc-1994-9-1023. PMID 7945680.
- ^ Biewenga, GP; Haenen, GRMM; Bast, A (1997). “Ch. 1: An Overview of Lipoate Chemistry”. In Fuchs, J; Packer, L; Zimmer, G (eds.). Lipoic Acid In Health & Disease. CRC Press. pp. 1–32. ISBN 9780824700935.
- ^ Lii, CK; Liu, KL; Cheng, YP; Lin, AH; et al. (May 2010). “Sulforaphane and alpha-lipoic acid upregulate the expression of the pi class of glutathione S-transferase through c-jun and Nrf2 activation”. Journal of Nutrition. 140 (5): 885–92. doi:10.3945/jn.110.121418. PMID 20237067.
- ^ Gal, EM; Razevska, DE (August 1960). “Studies on the in vivo metabolism of lipoic acid. 1. The fate of DL-lipoic acid-S35 in normal and thiamine-deficient rats”. Archives of Biochemistry and Biophysics. 89 (2): 253–61. doi:10.1016/0003-9861(60)90051-5. PMID 13825981.
- ^ Gal, EM (July 1965). “Reversal of selective toxicity of (-)-alpha-lipoic acid by thiamine in thiamine-deficient rats”. Nature. 207 (996): 535. Bibcode:1965Natur.207..535G. doi:10.1038/207535a0. PMID 5328673. S2CID 4146866.
- ^ US patent 6271254, Ulrich, H; Weischer, CH & Engel, J et al., “Pharmaceutical compositions containing R-alpha-lipoic acid or S-alpha.-lipoic acid as active ingredient”, issued 2001-08-07, assigned to ASTA Pharma.
- ^ Kilic, F; Handelman, GJ; Serbinova, E; Packer, L; et al. (October 1995). “Modelling cortical cataractogenesis 17: In vitro effect of a-lipoic acid on glucose-induced lens membrane damage, a model of diabetic cataractogenesis”. Biochemistry and Molecular Biology International. 37 (2): 361–70. PMID 8673020.
- ^ Artwohl, M; Schmetterer, L; Rainer, G; et al. (September 2000). Modulation by antioxidants of endothelial apoptosis, proliferation, & associated gene/protein expression. 36th Annual Meeting of the European Association for the Study of Diabetes, 17–21 September 2000, Jerusalem, Israel. Diabetologia. 43 (Suppl 1) (published August 2000). Abs 274. PMID 11008622.
- ^ Streeper, RS; Henriksen, EJ; Jacob, S; Hokama, JY; et al. (July 1997). “Differential effects of lipoic acid stereoisomers on glucose metabolism in insulin-resistant skeletal muscle”. AJP: Endocrinology and Metabolism. 273 (1 Pt 1): E185–91. doi:10.1152/ajpendo.1997.273.1.E185. PMID 9252495.
- ^ Frölich, L; Götz, ME; Weinmüller, M; Youdim, MB; et al. (March 2004). “(r)-, but not (s)-alpha lipoic acid stimulates deficient brain pyruvate dehydrogenase complex in vascular dementia, but not in Alzheimer dementia”. Journal of Neural Transmission. 111 (3): 295–310. doi:10.1007/s00702-003-0043-5. PMID 14991456. S2CID 20214857.
- ^ McIlduff, Courtney E; Rutkove, Seward B (2011-01-01). “Critical appraisal of the use of alpha lipoic acid (thioctic acid) in the treatment of symptomatic diabetic polyneuropathy”. Therapeutics and Clinical Risk Management. 7: 377–385. doi:10.2147/TCRM.S11325. ISSN 1176-6336. PMC 3176171. PMID 21941444.
- ^ Ziegle, D.; Reljanovic, M; Mehnert, H; Gries, F. A. (1999). “α-Lipoic acid in the treatment of diabetic polyneuropathy in Germany”. Experimental and Clinical Endocrinology & Diabetes. 107 (7): 421–30. doi:10.1055/s-0029-1212132. PMID 10595592.
- ^ “Lipoic Acid”. American Cancer Society. November 2008. Retrieved 5 October 2013.
- ^ Javed, S; Petropoulos, IN; Alam, U; Malik, RA (January 2015). “Treatment of painful diabetic neuropathy”. Therapeutic Advances in Chronic Disease. 6 (1): 15–28. doi:10.1177/2040622314552071. PMC 4269610. PMID 25553239.
- ^ Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF (April 2012). “Treatment for mitochondrial disorders”. Cochrane Database Syst Rev (4): CD004426. doi:10.1002/14651858.CD004426.pub3. PMC 7201312. PMID 22513923.
- ^ Namazi, Nazli; Larijani, Bagher; Azadbakht, Leila (2018). “Alpha-lipoic acid supplement in obesity treatment: A systematic review and meta-analysis of clinical trials”. Clinical Nutrition. 37 (2): 419–428. doi:10.1016/j.clnu.2017.06.002. ISSN 0261-5614. PMID 28629898
 |
|
 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
(R)-5-(1,2-Dithiolan-3-yl)pentanoic acid
|
|
| Other names
α-Lipoic acid; Alpha lipoic acid; Thioctic acid; 6,8-Dithiooctanoic acid
|
|
| Identifiers | |
|
|
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| DrugBank | |
| ECHA InfoCard | 100.012.793 |
| KEGG | |
| MeSH | Lipoic+acid |
|
PubChem CID
|
|
| UNII |
|
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C8H14O2S2 | |
| Molar mass | 206.32 g·mol−1 |
| Appearance | Yellow needle-like crystals |
| Melting point | 60–62 °C (140–144 °F; 333–335 K) |
| Very Slightly Soluble(0.24 g/L)[1] | |
| Solubility in ethanol 50 mg/mL | Soluble |
| Pharmacology | |
| A16AX01 (WHO) | |
| Pharmacokinetics: | |
| 30% (oral)[2] | |
| Related compounds | |
|
Related compounds
|
Lipoamide Asparagusic acid |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Infobox references | |
//////////Alpha lipoic acid, d-Thioctic acid, (R)-(+)-alpha-Lipoic acid, (R)-(+)-Thioctic acid, Dexlipotam,