Atosiban, sold under the brand name Tractocile among others, is an inhibitor of the hormones oxytocin and vasopressin. It is used as an intravenousmedication as a labour repressant (tocolytic) to halt premature labor. It was developed by Ferring Pharmaceuticals in Sweden and first reported in the literature in 1985.[5] Originally marketed by Ferring Pharmaceuticals, it is licensed in proprietary and generic forms for the delay of imminent preterm birth in pregnant adult women.
Atosiban is an inhibitor of the hormones oxytocin and vasopressin. It is used intravenously to halt premature labor. Although initial studies suggested it could be used as a nasal spray and hence would not require hospital admission, it is not used in that form. Atobisan was developed by the Swedish company Ferring Pharmaceuticals. It was first reported in the literature in 1985. Atosiban is licensed in proprietary and generic forms for the delay of imminent pre-term birth in pregnant adult women.
Medical uses
Atosiban is used to delay birth in adult women who are 24 to 33 weeks pregnant, when they show signs that they may give birth pre-term (prematurely).[4] These signs include regular contractions lasting at least 30 seconds at a rate of at least four every 30 minutes,[4] and dilation of the cervix (the neck of the womb) of 1 to 3 cm and an effacement (a measure of the thinness of the cervix) of 50% or more.[4] In addition, the baby must have a normal heart rate.[4]
Pharmacology
Mechanism of action
Atosiban is a nonapeptide, desamino-oxytocin analogue, and a competitive vasopressin/oxytocin receptor antagonist (VOTra). Atosiban inhibits the oxytocin-mediated release of inositol trisphosphate from the myometrial cell membrane. As a result, reduced release of intracellular, stored calcium from the sarcoplasmic reticulum of myometrial cells and reduced influx of Ca2+ from the extracellular space through voltage-gated channels occur. In addition, atosiban suppresses oxytocin-mediated release of PGE and PGF from the decidua.[6]
In human preterm labour, atosiban, at the recommended dosage, antagonises uterine contractions and induces uterine quiescence. The onset of uterus relaxation following atosiban is rapid, uterine contractions being significantly reduced within 10 minutes to achieve stable uterine quiescence.
Other uses
Atosiban use after assisted reproduction
Atosiban is useful in improving the pregnancy outcome of in vitro fertilization-embryo transfer (IVF-ET) in patients with repeated implantation failure.[7] The pregnancy rate improved from zero to 43.7%.[8]
First- and second-trimester bleeding was more prevalent in ART than in spontaneous pregnancies. From 2004 to 2010, 33 first-trimester pregnancies with vaginal bleeding after ART with evident uterine contractions, when using atosiban and/or ritodrine, no preterm delivery occurred before 30 weeks.[9]
In a 2010 meta-analysis,[10] nifedipine is superior to β2 adrenergic receptor agonists and magnesium sulfate for tocolysis in women with preterm labor (20–36 weeks), but it has been assigned to pregnancy category C by the U.S. Food and Drug Administration, so is not recommended before 20 weeks, or in the first trimester.[9] A report from 2011 supports the use of atosiban, even at very early pregnancy, to decrease the frequency of uterine contractions to enhance success of pregnancy.[7]
Pharmacovigilance
Following the launch of atosiban in 2000, the calculated cumulative patient exposure to atosiban (January 2000 to December 2005) is estimated as 156,468 treatment cycles. To date, routine monitoring of drug safety has revealed no major safety issues.[11]
Regulatory affairs
Atosiban was approved in the European Union in January 2000 and launched in the European Union in April 2000.[12][4] As of June 2007, atosiban was approved in 67 countries, excluding the United States and Japan.[12] It was understood that Ferring did not expect to seek approval for atosiban in the US or Japan, focusing instead on development of new compounds for use in Spontaneous Preterm Labor (SPTL).[12] The fact that atosiban only had a short duration before it was out of patent that the parent drug company decided not to pursue licensing in the US.[13]
Systematic reviews
In a systematic review of atosiban for tocolysis in preterm labour, six clinical studies — two compared atosiban to placebo and four atosiban to a β agonist — showed a significant increase in the proportion of women undelivered by 48 hours in women receiving atosiban compared to placebo. When compared with β agonists, atosiban increased the proportion of women undelivered by 48 hours and was safer compared to β agonists. Therefore, oxytocin antagonists appear to be effective and safe for tocolysis in preterm labour.[14]
A 2014 systematic review by the Cochrane Collaboration showed that while atosiban had fewer side effects than alternative drugs (such as ritodrine), other beta blockers, and calcium channel antagonists, it was no better than placebo in the major outcomes i.e. pregnancy prolongation or neonatal outcomes. The finding of an increase in infant deaths in one placebo-controlled trial warrants caution. Further research is recommended.[15]
PATENT
WO 2021207870
Atosiban (Atosiban) is an oxytocin and vasopressin V1A combined receptor antagonist, which can be used as a competitive antagonist of cyclic peptide oxytocin receptors in the uterus, decidua and fetal membrane. Atosiban is a disulfide-bonded cyclic polypeptide composed of 9 amino acids. It is a modified oxytocin molecule at positions 1, 2, 4 and 8. The N-terminal of the peptide is 3-mercaptopropionic acid (thiol and [ Cys] 6 thiol forms a disulfide bond), the C-terminal is in the form of an amide, and the second amino acid at the N-terminal is ethylated [D-Tyr(Et)] 2 . Atosiban is generally present in medicines in the form of acetate salt, commonly known as atosiban acetate. Its chemical formula is C 45 H 71 N 11 O 14 S 2 , its molecular weight is 994.19, and its structural formula is as follows:
[0003]
[0004]
In the prior art, atosiban is usually synthesized by a solid-phase peptide synthesis (SPPS) method, an amino resin is used as a starting carrier resin, and protected amino acids are sequentially connected, and the obtained atosiban is oxidized and then cleaved to obtain atosiban. However, the above-mentioned existing process has high cost, generates a large amount of solvent waste, and is not easy to monitor during the cyclization process. In addition, the above-mentioned prior art has deficiencies in the overall yield of crude peptides. Moreover, due to the existence of D-Tyr(Et) in the structure of atosiban, Fmoc-D-Tyr(Et) easily undergoes a racemization reaction during the peptide attachment process, resulting in [Tyr(Et) 2 ]-A The impurity of tosiban, which is similar in polarity to atosiban itself, is difficult to completely remove through purification, thus affecting the quality of atosiban.
[table 0001]
Amino acid name
alphabetic symbols
Glycine
Gly
Ornithine
Orn
Proline
Pro
cysteine
Cys
Asparagine
Asn
Threonine
Thr
Isoleucine
Ile
D-tyrosine (oxyethyl)
D-Tyr(ET)
Table 3 List of intermediates and Fmoc protected amino acids
According to the most preferred embodiment of the present invention, the method of the present invention comprises the following steps:
[0046]
The first step: Fmoc-Gly Rink resin can be directly purchased, which reduces the first step of synthesis and improves the synthesis efficiency;
[0047]
The second step: preparing a deprotection solution: the deprotection solution is a mixture of piperidine/N,N-dimethylformamide, preferably piperidine/N,N-dimethylformamide in a volume ratio of 1/4.
[0048]
The third step: preparation of Fmoc-Orn(Boc)-Gly Rink resin: deprotect the Fmoc-Gly Rink resin obtained in the first step, wash with DMF, add Fmoc-Orn(Boc)-OH in DMF solution, Condensation reaction is carried out under the condition of peptide coupling condensing agent to obtain Fmoc-Orn(Boc)-Gly Rink resin;
[0049]
The fourth step: preparation of Fmoc-Pro-Orn(Boc)-Gly Rink resin: the peptide resin obtained in the fourth step is deprotected and washed, and then reacted with Fmoc-Pro-OH under the condition of a peptide coupling agent to obtain Fmoc-Pro-Orn(Boc)-Gly Rink resin;
[0050]
The fifth step: preparation of Fmoc-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the fifth step is deprotected and washed, and then reacted with Fmoc-Cys(Trt)-OH under the condition of peptide coupling agent to obtain Fmoc-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0051]
The sixth step: preparation of Fmoc-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the sixth step is deprotected and washed, and then reacted with Fmoc-Asn-OH under the condition of peptide coupling agent to obtain Fmoc-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin ;
[0052]
The seventh step: preparation of Fmoc-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the seventh step was deprotected and washed, and then reacted with Fmoc-Thr(tBu)-OH under the condition of a peptide coupling agent. Obtain Fmoc-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0053]
The eighth step: preparation of Fmoc-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the eighth step is deprotected and washed, and then reacted with Fmoc-Ile-OH under the condition of a peptide coupling agent to obtain Fmoc-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn (Boc)-Gly Rink resin;
[0054]
The ninth step: preparation of Fmoc-D-Tyr(RT)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the ninth step is deprotected and washed, and then reacted with Fmoc-D-Tyr(ET)-OH under the condition of a peptide coupling agent to obtain Fmoc-D-Tyr(RT)-Ile-Thr(tBu )-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0055]
The tenth step: preparation of Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the tenth step is deprotected and washed, and then reacted with Mpa(Trt) under the condition of a peptide coupling agent to obtain Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn -Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0056]
The eleventh step: Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin in TFA/TIS/EDT/H2O =90/54/10/5 TFA, cleaved for 3 hours, and filtered to obtain crude peptide solution;
[0057]
The twelfth step: sedimentation and washing of the crude peptide solution with methyl tert-butyl ether, centrifugation at 2000 rpm, and vacuum drying to obtain a pale yellow solid powder of atosiban linear crude peptide;
[0058]
The thirteenth step: prepare three solutions for atosiban cyclization: solution A-sodium acetate buffered aqueous solution, solution B-aqueous solution of linear peptide atosiban crude peptide acetic acid, solution C: 30%-60% hydrogen peroxide solution ;
[0059]
The fourteenth step: Mix the above three solutions of A, B, and C at 15-25 ° C, and stir for 1-3 hours after mixing, so that the Mpa at the 1st position and the Cys at the 6th position form a disulfide bond to obtain Cyclized atosiban crude peptide.
[0060]
Step fifteen: Purify crude atosiban by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes.
[0061]
The sixteenth step: freeze-dry the purified atosiban solution at -50 to -70° C. for 18-48 hours with a freeze dryer.
[0062]
The purity of atosiban obtained by the method of the invention is more than 99.5%, and the total product yield is 55%-65%.
[0063]
The advantage of the method for preparing atosiban of the present invention is:
[0064]
The traditional SPPS synthesis of atosiban usually produces a large amount of waste with high disposal costs. This process adopts high-temperature SPPS process and selects different condensing agent combinations, which is faster than the conventional SPPS process, the product purity can reach more than 99.9%, the purity is better than that of the conventional atosiban process, the impurity content is low, and the product quality is high. The total yield can reach 55%-65%.
Detailed ways
[0065]
The invention will now be described with reference to specific embodiments. It must be understood that these examples are merely illustrative of the invention and are not intended to limit the scope of the invention. Unless otherwise stated, percentages and parts are by weight. Unless otherwise specified, experimental materials and reagents used in the following examples were obtained from commercial sources.
[0066]
Example 1:
[0067]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.61 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, other amino acids in the sequence are connected in the following order, and the coupling reagents are N,N-diisopropylcarbodiimide, 2-(7-benzotriazole)-N,N,N’,N ‘-Tetramethylurea hexafluorophosphate mixed in a volume ratio of 1:1, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 90°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated and washed twice with methyl tert-butyl ether, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 2:2 A solution-acetic acid-sodium acetate buffer aqueous solution (concentration is 30g/L), B solution-linear peptide atosiban crude peptide acetic acid aqueous solution and C solution: 60% hydrogen peroxide solution.
[0068]
The crude peptide yield was 85%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 18 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 56%.
[0069]
Example 2:
[0070]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, the other amino acids in the sequence are connected in the following order, and the coupling reagents are N,N-tert-dicyclohexylcarbodiimide, 1-hydroxybenzotriazole and Oxyma, which are mixed in a volume ratio of 1:1:1 , Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D- Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 90°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated with methyl tert-butyl ether and washed twice, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 3:2 solution A-formic acid-sodium formate buffer aqueous solution (concentration 25g/L), solution B-linear peptide atosiban crude peptide formic acid aqueous solution and solution C: 30% hydrogen peroxide solution, and oxygen was introduced.
[0071]
The crude peptide yield was 83%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 18 hours with a freeze dryer, the obtained atosiban has a purity greater than 99.5%, and the total product yield is 57%.
[0072]
Example 3:
[0073]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, other amino acids in the sequence were connected in the following order, and the coupling reagents were N,N-diisopropylethylamine, 2-(7-benzotriazole)-N,N,N’,N’- Two kinds of tetramethylurea hexafluorophosphate mixed in a 1:1 volume ratio, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc- Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 75°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated and washed twice with methyl tert-butyl ether, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 2:3 solution A-sodium phosphate buffered aqueous solution (concentration 15g/L), solution B-linear peptide atosiban crude peptide phosphoric acid aqueous solution and solution C: DMSO aqueous solution (volume 1:1).
[0074]
The crude peptide yield was 80%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70 DEG C for 28 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 55%.
[0075]
Example 4:
[0076]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:3 by volume). Subsequently, the other amino acids in the sequence were connected in the following order, and the coupling reagents were selected from 2-oxime ethyl cyanoacetate, N,N-diisopropylcarbodiimide, and 1-hydroxybenzotriazole in a volume ratio of 1. :1:1 mix, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH , Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 80°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated with methyl tert-butyl ether and washed twice, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 3:4 solution of A-trifluoroacetic acid-aqueous ammonia solution (concentration of 45 g/L), solution B-aqueous solution of linear peptide atosiban crude peptide trifluoroacetic acid and solution C: saturated aqueous iodine solution.
[0077]
The crude peptide yield was 78%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 38 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 52%.
Atosiban Acetate Injection was first listed in Austria on March 23, 2000 under the trade name: Atosiban, a new type of anti-prematurity drug developed by Ferring GmbH, which is an oxytocin The analog is a competitive antagonist of oxytocin receptors in the uterus, decidua, and fetal membranes. It is a first-line drug recommended by the European Medical Association; it can inhibit the binding of oxytocin and oxytocin receptors, thereby directly inhibiting the effect of oxytocin. In the uterus, it can inhibit uterine contraction; it can also inhibit the hydrolysis of phosphatidylinositol.
Atosiban is a cyclic nonapeptide whose molecular formula is C 43 H 67 N 11 O 12 S 2 ; molecular weight is 994.19; CAS registration number is 90779-69-4; its peptide sequence is as follows:
In the Chinese patents with announcement numbers CN101314613B and CN101696236B, the solid-phase synthesis of atosiban uses Rink Amide AM Resin resin solid-phase coupling stepwise to obtain Mpa(Trt)-D-Tyr(Et)-Ile-Thr(tBu)- Asn(Trt)-Cys(Trt)-Pro-Orn(Boc)-Gly-Resin is directly oxidized in solid phase to generate disulfide bonds, and then cleaved to obtain atosiban. The Rink Amide AM Resin resin used in the prior art needs to be cracked under a strong acid environment, which is not conducive to product stability and has a greater operational risk; Mpr and Cys both have sulfhydryl groups, and the sulfhydryl groups have the ability to capture tBu to generate double tBu impurities, When the peptide resin after solid-phase oxidation is cleaved to remove the protective group and resin, due to the presence of tBu or tBu source Boc protective group, it requires high capture agent, which is not conducive to product quality control and reduces product yield.
The Chinese patent with publication number CN105408344B discloses a method for synthesizing atosiban starting from Fmoc-Orn-Gly-NH2, wherein Fmoc-Orn-Gly-NH2 is connected to trityl through the side chain of ornithine On the base resin, impurities can be effectively controlled. However, using dipeptide and trityl-type resin for coupling, the resin attached to the Orn side chain of the dipeptide increases the steric hindrance of the subsequent Pro coupling and prolongs the coupling time, which is easy to cause missing peptide impurities.
Example 1. Synthesis of Fmoc-Pro-Orn-Gly-NH 2 tripeptide
[0027]
Fmoc-Pro-OH (134.94 g, 400 mmol) and N-hydroxysuccinimide (46.00 g, 400 mmol) were weighed into 1600 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (90.72g, 440mmol) in tetrahydrofuran (320ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 400 ml of tetrahydrofuran, and H-Orn(Boc)-NH 2 (92.92 g, 400 mmol) was dissolved in 300 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate to dryness under reduced pressure, add N-hydroxysuccinimide (46.00 g, 400 mmol) and 1600 ml of tetrahydrofuran to dissolve, and stir at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (90.72g, 440mmol) in tetrahydrofuran (320ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 400 ml of tetrahydrofuran, and H-Gly-NH 2 (29.64 g, 400 mmol) was dissolved in 300 ml of tetrahydrofuran and slowly added dropwise to the above solution, and the reaction was continued at room temperature after dropping, and the monitoring of the raw materials was completed. The reaction was filtered, and the filtrate was concentrated under reduced pressure. Dry, add 1000 mL of 5% TFA/DCM solution to the reaction solution, continue to react for 1 h, and concentrate to dryness to obtain a yellow oil, which is recrystallized from isopropanol to obtain 171.56 g of white solid with a yield of 69%.
[0028]
Example 2. Synthesis of Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.42 mmol/g
[0029]
Trityl resin (37.5 g, 30 mmol, substitution degree: 0.80 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 52.14 g of Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.42 mmol/g.
[0030]
Example 3. Synthesis of Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0031]
2-CTC Resin resin (30.0 g, 30 mmol, substitution degree: 1.00 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.80 g of Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0032]
Example 4. Synthesis of Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0033]
4-methyl-trityl resin (33.33 g, 30 mmol, substitution degree: 0.90 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.89 g of Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0034]
Example 5. Synthesis of Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0035]
4-Methoxy-trityl resin (30.0 g, 30 mmol, substitution degree: 1.00 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.69 g of Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0036]
Example 6. Synthesis of Atosiban Linear Peptide Resin 1
[0037]
Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 (35.71 g) prepared in Example 2 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0038]
Weigh 17.57g Fmoc-Cys(Trt)-OH and 4.86g HOBt, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.68g DIC (pre-cooled to <0°C), Activated in the solution for about 3 to 5 minutes, the activated solution was added to the reaction column under control, and reacted at 20 to 35 °C for 2 to 3 hours. The ninhydrin test was negative. The reaction solution was removed, and 200 mL of DMF was added to wash the resin. 6 times. After washing, the washing liquid was removed to obtain Fmoc-Cys(Trt)-Pro-Orn (trityl resin)-Gly-NH 2 .
[0039]
Repeat the step of receiving the peptide and remove the Fmoc protective group. According to the amino acid sequence of atosiban, Fmoc-Cys(Trt)-Pro-Orn (trityl resin)-Gly-NH 2 was coupled to Fmoc- Asn-OH, Fmoc-Thr-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(Et)-OH, Mpa(Trt)-OH give Mpa(Trt)-D-Tyr(Et)-Ile-Thr- Asn-Cys(Trt)-Pro-Orn (trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 48.72g after drying, and the resin weight gain was 89.0%.
[0040]
Example 7. Synthesis of atosiban linear peptide resin 2
[0041]
Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 (30.00 g) prepared in Example 3 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0042]
Weigh 17.57g Fmoc-Cys(Trt)-OH and 13.65g HBTU, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cooled to <0°C), put Activated in the solution for about 3 to 5 minutes, the activated solution was added to the reaction column under control, and reacted at 20 to 35 °C for 2 to 3 hours. The ninhydrin test was negative. The reaction solution was removed, and 200 mL of DMF was added to wash the resin. 6 times. After washing, the washing solution was removed to obtain Fmoc-Cys(Trt)-Pro-Orn(2-CTC Resin)-Gly-NH 2 .
[0043]
Fmoc-D-Tyr(Et)-OH (86.30 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and H-Ile-OH (26.24 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. The monitoring of the raw materials was completed. After filtration, the solution was concentrated under reduced pressure. , the concentrated solution was added to petroleum ether to separate out the solid, the solid was washed and then dried, recrystallized and dried with isopropanol to obtain 75.60 g of Fmoc-D-Tyr(Et)-Ile-OH with a yield of 75%.
[0044]
Repeat the step of receiving the peptide and removing the Fmoc protective group. According to the amino acid sequence of atosiban, sequentially couple Fmoc-Asn on Fmoc-Cys(Trt)-Pro-Orn(2-CTC Resin)-Gly-NH 2 -OH, Fmoc-Thr-OH, Fmoc-D-Tyr(Et)-Ile-OH, Mpa(Trt)-OH to give Mpa(Trt)-D-Tyr(Et)-Ile-Thr-Asn-Cys(Trt )-Pro-Orn( 2 -CTC Resin)-Gly-NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.77g after drying, and the resin weight gain rate was 87.4%.
[0045]
Example 8. Synthesis of atosiban linear peptide resin 3
[0046]
Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 (30.00 g) prepared in Example 4 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod, and the ninhydrin test was positive, indicating that Fmoc had been removed.
[0047]
Weigh 17.57g Fmoc-Cys(Trt)-OH, 13.65g HBTU and 4.05g HOBt, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cooled to <0 ℃), activate in the solution for about 3-5min, add the activated solution to the reaction column, react at 20-35 ℃ for 2-3h, the ninhydrin test is negative, remove the reaction solution, add 200mL of DMF The resin was washed 6 times. After washing, the washing liquid was removed to obtain Fmoc-Cys(Trt)-Pro-Orn(4-methyl-trityl resin)-Gly-NH 2 .
[0048]
Mpa(Trt)-OH (69.69 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and HD-Tyr(Et)-OH (41.85 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate under reduced pressure, add the concentrated solution to petroleum ether to precipitate a solid, wash the solid and then dry it, recrystallize and dry with isopropanol to obtain Mpa(Trt)-D-Tyr(Et)-OH 77.98g, yield 72%.
[0049]
Repeat the step of receiving the peptide and removing the Fmoc protective group, according to the amino acid sequence of atosiban, on Fmoc-Cys(Trt)-Pro-Orn (4-methyl-trityl resin)-Gly- NH 2 Fmoc-Asn-OH, Fmoc-Thr-OH, Fmoc-Ile-OH, Mpa(Trt)-D-Tyr(Et)-OH were sequentially coupled to obtain Mpa(Trt)-D-Tyr(Et)-Ile-Thr -Asn-Cys(Trt)-Pro-Orn(4-methyl-trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.91g after drying, and the resin weight gain rate was 88.3%.
[0050]
Example 9. Synthesis of atosiban linear peptide resin 4
[0051]
Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 (30.00 g) prepared in Example 5 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0052]
Fmoc-Asn-OH (70.87 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and H-Cys(Trt)-OH (79.96 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate under reduced pressure, add the concentrated solution to petroleum ether to precipitate a solid, wash the solid and then dry, recrystallize and dry with isopropanol to obtain Fmoc-Asn-Cys(Trt)-OH 102.17g, yield 73%.
[0053]
Weigh 20.99g Fmoc-Asn-Cys(Trt)-OH and 13.65g HCTU, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cool to <0°C) , activate in the solution for about 3-5min, add the activated solution to the reaction column, react at 20-35°C for 2-3h, the ninhydrin test is negative, remove the reaction solution, add 200mL of DMF to wash the resin , wash 6 times. After washing, the washing liquid was removed to obtain Fmoc-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly-NH 2 .
[0054]
Repeat the step of receiving the peptide and removing the Fmoc protective group. According to the amino acid sequence of atosiban, in Fmoc-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly- Fmoc-Thr-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(Et)-OH, Mpa(Trt)-OH were sequentially coupled on NH 2 to obtain Mpa(Trt)-D-Tyr(Et)-Ile- Thr-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.28g after drying, and the resin weight gain was 84.0%.
[0055]
Example 10. Synthesis of atosiban crude peptide 1
[0056]
Configure 487.2ml of TFA/DCM=2/98 (V/V) lysis solution, cool to 5-10°C, add 48.72g of peptide resin prepared in Example 6 into the lysis solution, at room temperature (20-35°C) React for 5h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.77g of atosiban linear peptide, dissolve 14.30g of atosiban linear peptide in 0.75L of glacial acetic acid, add 6.75L of water to dilute, add 0.1M/L iodine ethanol solution dropwise until the solution changes color, react at room temperature for 1.0h, That is, the crude atosiban peptide is obtained, and its HPLC spectrum is shown in Figure 1.
[0057]
Example 11. Synthesis of atosiban crude peptide 2
[0058]
Configure TFA/DCM=5/95 (V/V) lysate 448.6ml, cool to 5~10℃, add 42.77g of peptide resin prepared in Example 7 into the lysate, at room temperature (20~35℃) React for 3h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate, dry to obtain a solid, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.21g of atosiban linear peptide, dissolve 14.21g of atosiban linear peptide in 1.5L of glacial acetic acid, add 6L of water to dilute, add 0.1M/L iodoethanol solution dropwise until the solution changes color, react at room temperature for 1.0h, that is The crude atosiban peptide was obtained, and its HPLC chromatogram was similar to that in Figure 1.
[0059]
Example 12. Synthesis of atosiban crude peptide 3
[0060]
Configure 450.5ml of TFA/DCM=20/80(V/V) lysis solution, cool to 5~10℃, add 45.05g of peptide resin prepared in Example 8 into the lysis solution, at room temperature (20~35℃) React for 2h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.63g of atosiban linear peptide, dissolve 14.63g of atosiban linear peptide in 1.5L of glacial acetic acid, add 6L of water to dilute, add 10% hydrogen peroxide solution, and react at room temperature for 1.0h to obtain atosiban Crude peptide, its HPLC chromatogram is similar to Figure 1.
[0061]
Example 13. Synthesis of atosiban crude peptide 4
[0062]
Configure TFA/DCM=1/99 (V/V) lysate 442.7ml, cool to 5~10℃, add 44.27g of peptide resin prepared in Example 9 into the lysate, at room temperature (20~35℃) React for 5h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.13 g of atosiban linear peptide, dissolve 14.13 g of atosiban linear peptide in 1.5 L of glacial acetic acid, add 6 L of water to dilute, add 30% hydrogen peroxide solution, and react at room temperature for 1.0 h to obtain atosiban Crude peptide, its HPLC chromatogram is similar to Figure 1.
[0063]
Example 14. Purification of atosiban crude peptide 1
[0064]
The atosiban crude peptide prepared in Example 10 was dissolved in 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.12g , the yield is 64%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is shown in Figure 2.
[0065]
Example 15. Purification of atosiban crude peptide 2
[0066]
The crude atosiban peptide obtained in Example 11 was dissolved in a 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 9.80 g , the yield is 62%, the purity is 99%, and the obtained atosiban peptide HPLC spectrum is similar to Figure 2.
[0067]
Example 16. Purification of atosiban crude peptide 3
[0068]
The crude atosiban peptide obtained in Example 12 was dissolved in a 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.28g , the yield is 65%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is similar to that in Figure 2.
[0069]
Example 17. Purification of atosiban crude peptide 4
[0070]
The crude atosiban peptide obtained in Example 13 was dissolved in 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.27g , the yield is 65%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is similar to that in Figure 2.
Atosiban is a nonapeptide which contains three non-natural amino acids: D-Tyr(Et), Mpa and Orn, and a pair of disulfide bonds looped between Mpa and Cys, the structural formula is: c[Mpa-D-Tyr(Et)-Ile-Thr-Asn-Cys]-Pro-Orn-Gly-NH2.
By means of competing for oxytocin receptor with oxytocin, Atosiban can inhibit the combination between oxytocin and oxytocin receptor, and directly prevent the oxytocin from acting on uterus, and then inhibit the uterine contraction; as another hand, atosiban can also inhibit the hydrolysis of phosphatidylinositol and then block the generation of messenger and activity of Ca2+, with the decreasing of activity from oxytocin, the contraction of uterine is indirectly inhabited.
At present, there are many reports about synthesis process method in China and abroad A report in China shows that the inventor found a simple process by adopting solid phase oxidation, resulting in a low purity crude product, with low yield and low application value. The aforementioned reports about atosiban synthesis process reveal that most of them adopt the method using Boc solid phase synthetic and cleaving peptide with liquid ammonia, then oxidating with liquid phase oxidation, and purifying. Those respective processes result in “the three wastes” and are too complex for industrial production. See U.S. Pat. No. 4,504,469.
Example 1Preparing the Linear Atosiban Peptide Resin
(i) 6.25 g of Rink Amide resin (substitutability=0.8 mmol/g) is put into a reaction bottle, DMF is added into the bottle and washed twice, then swelled for 30 min with DMF. Fmoc protecting group of Rink Amide resin is removed with 30-40 ml of 20% DBLK, washed for 4 times with DMF, then washed twice with DCM after removal, the product is detected by ninhydrin detecting method, the resin is reddish-brown.
(ii) 4.46 g of Fmoc-Gly-OH and 2.43 g of HOBt dissolved in a suitable amount of DMF, which had been pre-activated with 3.05 ml DIC; the mixture is, added to the reaction bottle, and reacted for 2 h, the resin is negative by ninhydrin detecting method, after the reaction, the product is washed for 4 times with DMF, then washed twice with DCM, if the resin is positive, repeating the above condensation reaction until negative.
(iii) Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH and Mpa(Trt)-OH are coupled orderly.
Example 2Cleaving the Linear Atosiban Peptide Resin
5.15 g of linear atosiban is prepared by washing the linear atosiban peptide resin obtained from Example 1 for 3 times with 30 ml of methanol, adding the dry resin obtained to 150 ml of mixed solution with a volume ratio of TFA:H2O=95:5, reacting for 2 hours at 25° C. and filtering, washing the resin for 3 times with few trifluoroacetic acid, combining the filtrate and pouring into 1500 ml glacial ether, making rest for 2 hours, centrifugally separating the linear atosiban, washing for 3 times, and drying in a vacuum drier, MS: 995.3, HPLC: 91.5%, content: 65.5%, synthesis yield: 68%.
Example 3Oxidizing the Linear Atosiban
2.85 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 5% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.60 g of H2O2, reacting for 10 min at 25° C., monitoring with HPLC (HPLC: 75.6%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.4%.
Example 4Oxidizing the Linear Atosiban
3.01 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 10% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.85 g of H2O2, reacting for 30 min at 25° C., monitoring with HPLC (HPLC: 89.5%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.6%.
Example 5Oxidizing the Linear Atosiban
2.95 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 10% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.85 g of H2O2, reacting for 60 min at 25° C., monitoring with HPLC (HPLC: 83.5%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.4%.
The above is the further detailed description of the invention in conjunction with specific preferred examples, but it should not be considered that the specific examples of the invention are only limited to the these descriptions. For one of ordinary skill in the art, many deductions and replacements can be made without departing from the inventive concept. Such deductions and replacements should fall within the scope of protection of the invention.
Clips
https://www.mdpi.com/1420-3049/27/6/1920/htm
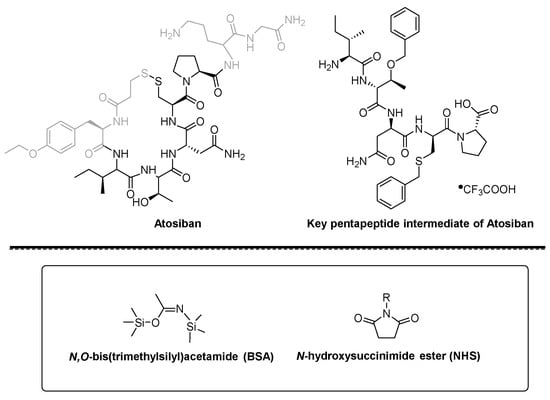
Figure 1. Structure of Atosiban, pentapeptide intermediate, BSA and NHS ester.
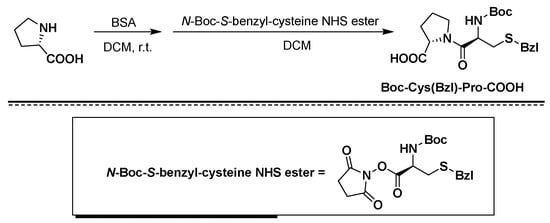
Figure 2. Synthesis of Boc-Cys(Bzl)-Pro-COOH using BSA/NHS as coupling agents.
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
A 2013 retrospective study comparing the efficacy and safety of atosiban and nifedipine in the suppression of preterm labour concluded that atosiban and nifedipine are effective in delaying delivery for seven days or more in women presenting with preterm labour.[16] A total of 68.3% of women in the atosiban group remained undelivered at seven days or more, compared with 64.7% in the nifedipine group.[16] They have the same efficacy and associated minor side effects.[16] However, flushing, palpitation, and hypotension were significantly higher in the nifedipine group.[16]
A 2012 clinical trial compared tocolytic efficacy and tolerability of atosiban with that of nifedipine.[17] Forty-eight (68.6%) women allocated to atosiban and 39 (52%) to nifedipine did not deliver and did not require an alternate agent at 48 hours, respectively (p=.03).[17] Atosiban has fewer failures within 48 hours.[17] Nifedipine may be associated with a longer postponement of delivery.[17]
A 2009 randomised controlled study demonstrated for the first time the direct effects of atosiban on fetal movement, heart rate, and blood flow.[18] Tocolysis with either atosiban or nifedipine combined with betamethasone administration had no direct fetal adverse effects.[18]
Atosiban vs. ritodrine
Multicentre, controlled trial of atosiban vs. ritodrine in 128 women shows a significantly better tocolytic efficacy after 7 days in the atosiban group than in the ritodrine group (60.3 versus 34.9%), but not at 48 hours (68.3 versus 58.7%). Maternal adverse events were reported less frequently in the atosiban group (7.9 vs 70.8%), resulting in fewer early drug terminations due to adverse events (0 versus 20%). Therefore, atosiban is superior to ritodrine in the treatment of preterm labour.[19]
Brand names
In India it is marketed under the brand name Tosiban by Zuventus healthcare ltd.
^Akerlund M, Carlsson AM, Melin P, Trojnar J (1985). “The effect on the human uterus of two newly developed competitive inhibitors of oxytocin and vasopressin”. Acta Obstet Gynecol Scand. 64 (6): 499–504. doi:10.3109/00016348509156728. PMID4061066. S2CID25799128.
^Lamont, Ronald F; Kam, KY Ronald (March 2008). “Atosiban as a tocolytic for the treatment of spontaneous preterm labor”. Expert Review of Obstetrics & Gynecology. 3 (2): 163–174. doi:10.1586/17474108.3.2.163. ISSN1747-4108.
^ Jump up to:abcLamont, Ronald F.; Kam, KY Ronald (2008). “Atosiban as a tocolytic for the treatment of spontaneous preterm labor”. Expert Review of Obstetrics & Gynecology. 3 (2): 163–174. doi:10.1586/17474108.3.2.163.
^Lamont CD, Jørgensen JS, Lamont RF (September 2016). “The safety of tocolytics used for the inhibition of preterm labour”. Expert Opinion on Drug Safety. 15 (9): 1163–73. doi:10.1080/14740338.2016.1187128. PMID27159501. S2CID4937942. It was for this reason and the fact that Tractocile (atosiban) only had a short duration before it was out of patent that the parent drug company decided not to pursue licensing in the USA.
^Coomarasamy, A; Knox, EM; Gee, H; Khan, KS (November 2002). “Oxytocin antagonists for tocolysis in preterm labour — a systematic review”. Med Sci Monit. 8 (11): RA268–73. PMID12444392.
^ Jump up to:abde Heus R, Mulder EJ, Derks JB, Visser GH (June 2009). “The effects of the tocolytics atosiban and nifedipine on fetal movements, heart rate and blood flow”. J Matern Fetal Neonatal Med. 22 (6): 485–90. doi:10.1080/14767050802702349. PMID19479644. S2CID35810758.
^Shim JY, Park YW, YoonBH, Cho YK, Yang JH, Lee Y, Kim A. “Multicentre, parallelgroup, randomised, single-blind study of the safety and efficacy of atosibanversus ritodrine in the treatment of acute preterm labour in Korean women. BJOG 2006Nov;113(11):1228-34.
External links
“Atosiban”. Drug Information Portal. U.S. National Library of Medicine.





















 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.