














BMS-791325, Beclabuvir
IN PHASE 2 for Hepatitis C (HCV)
An NS5B inhibitor.

BMS-791325 preferably is


958002-33-0
958002-36-3 (as hydrochloride)
C36 H45 N5 O5 S, 659.838
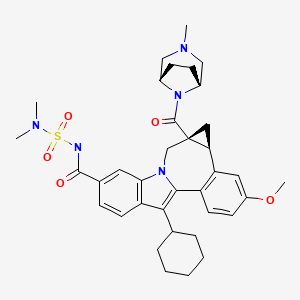
Cycloprop(d)indolo(2,1-a)(2)benzazepine-9-carboxamide, 12-cyclohexyl-N-((dimethylamino)sulfonyl)-4b,5,5a,6-tetrahydro-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-, (4bS,5aR)-
(4bS,5aR)-12-Cyclohexyl-N-(dimethylsulfamoyl)-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-4b,5,5a,6-tetrahydrocyclopropa(d)indolo(2,1-a)(2)benzazepine-9-carboxamide
(4bS,5aR)-12-Cyclohexyl-N-(dimethylsulfamoyl)-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-4b,5,5a,6-tetrahydrocyclopropa(d)indolo(2,1-a)(2)benzazepine-9-carboxamide
(1aR,12bS)-8-Cyclohexyl-N-(dimethylsulfamoyl)-11-methoxy-1a-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8-yl)carbonyl]-1,1a,2,12b-tetrahydrocyclopropa[d]indolo[2,1-a][2]benzazepine-5-carboxamide
Cycloprop [d] indolo [2, 1 -a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (laR,12bS)-
Bristol-Myers Squibb (Originator)
RNA-Directed RNA Polymerase (NS5B) Inhibitors
UNII-MYW1X5CO9S
BMS-791325 is in phase II clinical studies at Bristol-Myers Squibb for the treatment of chronic hepatitis C. In 2013, the company received breakthrough therapy designation in the U.S. for the treatment of chronic hepatitis C in combination with daclatasvir and asunaprevir.
| Patent | WO 2007136982 |
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
http://www.google.com/patents/WO2007136982A1?cl=en
Scheme 1.

N-protected piperazines can also be coupled to the intermediate indolobenzazepine acids and the resultant piperazine carboxamides can be deprotected using methods known in the art and derivatized using a variety of synthetic protocols, some illustrative examples of which are shown below (See Scheme 2).
Scheme 2.

An intermediate useful for the synthesis of some compounds of the invention involves the preparation of the tert-butyl ester indolobenzazepine shown in Scheme 3. Scheme 3.

t-Butylation either:

This methodology involves base catalyzed hydrolysis of the indole methyl ester shown, followed by its reaction with either thionyl chloride and potassium tertiary butoxide, or alkylation with silver carbonate and tertiary butyl bromides. The resultant compound can be transformed using chemistry analogous to that outlined previously to provide the mixed ester indolobenzazepines shown above.
Scheme 4.

Some examples exist as stereoisomeric mixtures. The invention encompasses all stereoisomers of the compounds. Methods of fractionating stereoisomeric mixtures are well known in the art, and include but are not limited to; preparative chiral supercritical fluid chromatography (SFC) and chiral high performance liquid chromatography (HPLC). An example using this approach is shown in scheme 5. Scheme 5.

An additional method to achieve such separations involves the preparation of mixtures of diastereomers which can be separated using a variety of methods known in the art. One example of this approach is shown below (Scheme 6).
Scheme 6.

Diastereomers separated by reverse phase HPLC
Some diastereomeric amides can be separated using reverse phase HPLC. After hydroysis, the resultant optically active acids can be coupled with bridged piperazine derivatives (Scheme 6). For example, O-(lH-benzotriazol-l-yl)-N,N, N’,N’-tetramethyluronium tetrafluoroborate and diisopropyl ethyl amine in DMSO can be used to give the alkyl bridged piperazine carboxamides. Other standard acid amine coupling methods can also be used to give optically active carboxamides.

Schemes 7-9 illustrate other methods of making intermediates and compounds.

Scheme 8.

Scheme 9.


Biological Methods
The compounds demonstrated activity against HCV NS5B as determined in the following HCV RdRp assays.
DESCRIPTION OF SPECIFIC EMBODIMENTS
Unless otherwise specified, analytical LCMS data on the following intermediates and examples were acquired using the following columns and conditions. Stop time: Gradient time + 1 minute; Starting cone: 0% B unless otherwise noted; Eluent A: 5% CH3CN / 95% H2O with 10 mM NH4OAc (for columns A, D and E); 10 % MeOH / 90 % H2O with 0.1% TFA (for columns B and C); Eluent B: 95% CH3CN / 5% H2O with 10 mM NH4OAc (for columns A, D and E); 90 % MeOH / 10 % H2O with 0.1% TFA (for columns B and C); Column A:
Phenomenex lOμ 4.6 x 50 mm C18; Column B: Phenomenex C18 lOμ 3.0 x 50 mm; Column C: Phenomenex 4.6 x 50 mm C18 lOμ; Column D: Phenomenex Lina C18 5μ 3.0 x 50 mm; Column E: Phenomenex 5μ 4.6 x 50 mm Cl 8.
Intermediate 1

lH-Indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-, methyl ester. Freshly recrystallized pyridinium tribromide (recrystallization from hot AcOH (5 mL per 1 g), rinsed with cold AcOH and dried under high vacuum over KOH) was added in portions (over 10 min.) to a stirring solution of methyl 3-cyclohexyl-lH-indole-6- carboxylate (60 g, 233 mmol) (prepared using procedures describe in WO2004/065367) in CHC1/THF (1: 1, 1.25 L) at 2o C. The reaction solution was stirred at 0-5 °C for 2.5h, and washed with sat. aq. NaHSO3 (1 L), 1 N HCl (1 L) and brine (1 L). The organic layer was dried (MgSO4) and concentrated. The resulting red oil was diluted with Et2θ and concentrated. The resulting pink solid was dissolved into Et2θ (200 mL) treated with hexanes (300 mL) and partially concentrated. The solids were collected by filtration and rinsed with hexanes. The mother liquor was concentrated to dryness and the procedure repeated. The solids were combined to yield lH-indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-, methyl ester (64 g, 190 mmol, 82%) as a fluffy pink solid, which was used without further purification. IHNMR (300 MHz, CDCl3) δ 8.47 (br s, IH), 8.03 (d, J = 1.4 Hz, IH), 7.74 (dd, J = 1.4, 8.8 Hz, IH), 7.69 (d, J = 8.8 Hz, IH), 3.92 (s, 3H), 2.82 (tt, J = 3.7, 11.7 Hz, IH), 1.98 – 1.72 (m, 7H), 1.50 – 1.27 (m, 3H). 13CNMR (75 MHz, CDC13) δ 168.2, 135.6, 130.2, 123.1, 120.8, 120.3, 118.7, 112.8, 110.7, 52.1, 37.0, 32.2(2), 27.0(2), 26.1. LCMS: m/e 334 (M-H)“, ret time 3.34 min, column A, 4 minute gradient.
Intermediate 2

lH-Indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-. A solution of methyl 2- bromo-S-cyclohexyl-lH-indole-ό-carboxylate (20 g, 60 mmol) and LiOH (3.8 g, 160 mmol) in MeOΗ/TΗF/Η2O ( 1 : 1 : 1 , 300 mL) was heated at 90 °C for 2h. The reaction mixture was cooled in an ice/H2O bath, neutralized with IM HCl (-160 mL) diluted with H2O (250 mL) and stirred for Ih at rt. The precipitates were collected by filtration rinse with H2O and dried to yield lH-indole-6-carboxylic acid, 2-bromo-3- cyclohexyl- (quant.) which was used without further purification.
An alternative procedure that can by used to provide lH-indole-6-carboxylic acid, 2-bromo-3-cyclohexyl- is described below: A solution of methyl 2-bromo-3-cyclohexyl-lH-indole-6-carboxylate (117 g, 349 mmol) and LiOKH2O (26.4 g, 629 mmol) in MeOH/THF/H2O (1: 1: 1, 1.8 L) was heated at reflux for 3h. The reaction mixture was cooled in an ice/H2O bath to ~2 °C, neutralized with IM HCl (-650 mL) (added at such a rate that temperature did not exceed 5 °C), diluted with H2O (1 L) and stirred while warming to ambient temperature. The precipitates were collected by filtration rinsed with H2O and dried to yield the mono THF solvate of lH-indole-6-carboxylic acid, 2-bromo-3- cyclohexyl- (135.5 g, 345 mmol, 99%) as a yellow solid, which was used without further purification. IHNMR (300 MHz, CDCl3) δ 11.01 (br s, IH), 8.77 (s, IH), 8.07 (d, J = 1.5 Hz, IH), 7.82 (dd, J = 1.5, 8.8 Hz, IH), 7.72 (d, J = 8.8 Hz, IH), 3.84 – 3.74 (m, 4H), 2.89 (m, IH), 1.98 – 1.72 (m, HH), 1.50 – 1.24 (m, 3H). 13CNMR (75 MHz, CDC13) δ 172.7, 135.5, 130.7, 122.3, 120.9(2), 118.8, 113.3, 111.1, 67.9(2), 37.0, 32.2(2), 27.0(2), 26.1, 25.5(2). LCMS: m/e 320 (M-H)“, ret time 2.21 min, column A, 4 minute gradient.
Intermediate 3

lH-Indole-6-carboxamide, 2-bromo-3-cyclohexyl-N-
[(dimethylamino)sulfonyl]-. l,l’-Carbonyldiimidazole (1.17 g, 7.2 mmol) was added to a stirred solution of 2-bromo-3-cyclohexyl-lH-indole-6-carboxylic acid (2.03 g, 6.3 mmol) in THF (6 mL) at 22 °C. The evolution of CO2 was instantaneous and when it slowed the solution was heated at 50°C for 1 hr and then cooled to 220C. N,N-Dimethylsulfamide (0.94 g, 7.56 mmol) was added followed by the dropwise addition of a solution of DBU (1.34 g ,8.8 mmol) in THF (4 mL). Stirring was continued for 24 hr. The mixture was partitioned between ethyl acetate and dilute HCl. The ethyl acetate layer was washed with water followed by brine and dried over Na2SO4. The extract was concentrated to dryness to leave the title product as a pale yellow friable foam, (2.0 g, 74 %, >90 % purity , estimated from NMR). 1H NMR (300 MHz, DMSO-D6) δ ppm 1.28 – 1.49 (m, 3 H) 1.59 – 2.04 (m, 7 H) 2.74 – 2.82 (m, 1 H) 2.88 (s, 6 H) 7.57 (dd, J=8.42, 1.46 Hz, 1 H) 7.74 (d, J=8.78 Hz, 1 H) 7.91 (s, 1 H) 11.71 (s, 1 H) 12.08 (s, 1 H).
An alternative method for the preparation of lH-indole-6-carboxamide, 2- bromo-3-cyclohexyl-N-[(dimethylamino)sulfonyl]- is described below.
To a 1 L four necked round bottom flask equipped with a mechanical stirrer, a temperature controller, a N2 inlet , and a condenser, under N2, was added 2-bromo-3- cyclohexyl-lH-indole-6-carboxylic acid (102.0 g, 0.259 mol) and dry TΗF (300 mL). After stirring for 10 min, CDI (50.3 g, 0.31 mol) was added portion wise. The reaction mixture was then heated to 50 oC for 2 h. After cooling to 30 oC, N,N- dimethylaminosulfonamide (41.7 g, 0.336 mol) was added in one portion followed by addition of DBU (54.1 mL, 0.362 mol) drop wise over a period of 1 h. The reaction mixture was then stirred at rt for 20 h. The solvent was removed in vacuo and the residue was partitioned between EtOAc and 1 Ν HCl (1 : 1, 2 L). The organic layer was separated and the aqueous layer was extracted with EtOAc (500 mL). The combined organic layers were washed with brine (1.5 L) and dried over MgSO4. The solution was filtered and concentrated in vacuo to give the crude product (111.0 g). The crude product was suspended in EtOAc (400 mL) at 60 oC. To the suspension was added heptane (2 L) slowly. The resulting suspension was stirred and cooled to 0 oC. It was then filtered. The filter cake was rinsed with small amount of heptane and house vacuum air dried for 2 days. The product was collected as a white solid (92.0 g, 83%). 1H ΝMR (MeOD, 300 MHz) δ 7.89 (s, H), 7.77 (d, J= 8.4 Hz, IH), 7.55 (dd, J= 8.4 and 1.8 Hz, IH), 3.01 (s, 6H), 2.73-2.95 (m, IH), 1.81-2.05 (m, 8H), 1.39-1.50 (m, 2H); m/z 429 (M +H)+. Intermediate 4

lH-Indole-6-carboxamide, 3-cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2- formyl-4-methoxyphenyl)-. A mixture of the 2-Bromo-3-cyclohexyl- N- [(dimethylamino)sulfonyl]-lH-indole-6-carboxamide (4.28g, 0.01 mol), 4-methoxy- 2-formylphenyl boronic acid (2.1%, 0.015 mol), 2-dicyclohexylphosphino-2′,6′- dimethoxy-biphenyl (41 mg, 0.0001 mol), palladium acetate (11.2 mg), and finely ground potassium carbonate (4.24g, 0.02 mol) in toluene (30 mL) was stirred under reflux and under nitrogen for 30 min, at which time LC/MS analysis showed the reaction to be complete. The reaction mixture was then diluted with ethyl acetate and water, and then acidified with an excess of dilute HCl. The ethyl acetate layer was then collected and washed with dilute HCl, water and brine. The organic solution was then dried (magnesium sulfate), filtered and concentrated to give a gum. The gum was diluted with hexanes (250 ml) and ethyl acetate (25 mL), and the mixture was stirred for 20 hr at 22° C during which time the product was transformed into a bright yellow granular solid (4.8 g) which was used directly without further purification.
An alternative procedure for the preparation of lH-indole-6-carboxamide, 3- cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2-formyl-4-methoxyphenyl)- is provided below:
To a slurried solution of 2-bromo-3-cyclohexyl-N-[(dimethylamino)sulfonyl]- indole-6-carboxamide (54.0 g, 126 mmol), 4-methoxy-2-formylphenylboronic acid (29.5 g, 164 mmol) and LiCl (13.3 g, 315 mmol) in EtOH/toluene (1 : 1, 1 L) was added a solution of Na2CO3 (40.1 g, 379 mmol) in water (380 mL). The reaction mixture was stirred 10 min. and then Pd(PPh3)4 (11.3 g, 10.0 mmol) was added. The reaction solution was flushed with nitrogen and heated at 70 °C (internal monitoring) overnight and then cooled to rt. The reaction was diluted with EtOAc (1 L) and EtOH (100 mL), washed carefully with IN aqueous HCl (1 L) and brine (500 mL), dried (MgSO4), filtered and concentrated. The residual solids were stirred with Et20 (600 mL) for Ih and collected by filtration to yield lH-indole-6-carboxamide, 3- cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2-formyl-4-methoxyphenyl)- (52.8g, 109 mmol, 87%) as a yellow powder which was used without further purification. IHNMR (300 MHz, d6-DMSO) δ 11.66 (s, IH), 8.17 (s, IH), 7.75 (d, J = 8.4 Hz, IH), 7.74 (d, J = 8.4 Hz, IH), 7.59 (dd, J = 1.4, 8.4 Hz, IH), 7.23 – 7.16 (m, 2H), 7.08 (dd, J = 2.6, 8.4 Hz, IH), 6.54 (d, J = 8.8 Hz, IH), 3.86 (s, 3H), 3.22 – 3.08 (m, IH), 2.91 (s, 6H), 2.00 – 1.74 (m, 7H), 1.60 – 1.38 (m, 3H). 13CNMR (75 MHz, CDC13) δ 165.7, 158.8, 147.2, 139.1, 134.3, 132.0, 123.4, 122.0, 119.2, 118.2, 114.8, 112.3, 110.4, 109.8, 79.6, 45.9, 37.2(2), 34.7, 32.0(2), 25.9(2), 24.9. LCMS: m/e 482 (M- H)“, ret time 2.56 min, column A, 4 minute gradient.
Intermediate 5

6H-Isoindolo[2,l-a]indole-3-carboxamide, 11-cyclohexyl-N-
[(dimethylamino)sulfonyl]-6-ethoxy-8-methoxy-. To a 5 L four necked round bottom flask equipped with a temperature controller, a condenser, a N2 inlet and a mechanical stirrer, was charged toluene (900 mL), EtOH (900 mL), 2-bromo-3- cyclohexyl-N^NjN-dimethylsulfamoyiyiH-indole-ό-carboxamide (90 g, 0.21 mol), 2-formyl-4-methoxyphenylboronic acid (49.2 g, 0.273 mol) and LiCl (22.1 g, 0.525 mol). The resulting solution was bubbled with Ν2 for 15 mins. A solution of Na2CO3 (66.8 g, 0.63 mol) in Η2O (675 mL) was added and the reaction mixture was bubbled with N2 for another (10 mins). Pd(PPh3)4 (7.0 g, 6.3 mmol) was added and the reaction mixture was heated to 70 °C for 20 h. After cooling to 35 °C, a solution of 1 N HCl (1.5 L) was added slowly. The resulting mixture was transferred to a 6 L separatory funnel and extracted with EtOAc (2 X 1.5 L). The combined organic extracts were washed with brine (2 L), dried over MgSO4, filtered and concentrated in vacuo to give a yellow solid, which was triturated with 20% EtOAc in hexane (450 mL, 50 °C to 0 °C) to give 3-cyclohexyl-N-(N,N-dimethylsulfamoyl)-2-(2-formyl-4- methoxyphenyl)-lH-indole-6-carboxamide(65.9 g) as a yellow solid. HPLC purity, 98%.
The mother liquid from the trituration was concentrated in vacuo. The residue was refluxed with EtOH (50 mL) for 3 h. The solution was then cooled to 0 °C. The precipitates were filtered and washed with cooled TBME (5 °C) (20 mL). The filter cake was house vacuum air dried to give a further quantity of the title compound as a white solid (16.0 g). HPLC purity, 99%. 1H NMR (CDC13, 300 MHz) δ 8.75 (s, IH), 7.96 (s, IH), 7.73 (d, J= 8.4 Hz, IH), 7.67 (d, J= 8.4 Hz, IH), 7.45 (dd, J= 8.4 and 1.4 Hz, IH), 7.09 (d, J= 2.2 Hz, IH), 6.98 (dd, J= 8.4 and 2.2 Hz, IH), 6.50 (s, IH), 3.86 (s, 3H), 3.05 (s, 6H), 2.92-3.13 (m, 3H), 1.85-1.93 (m, 7 H), 1.40-1.42 (m, 3H), 1.05 (t, J= 7.1 Hz, 3H). m/z 512 (M + H)+.
Intermediate 6

lH-indole-6-carboxamide, 3-cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2- formyl-4-methoxyphenyl)-. 1 l-cyclohexyl-N-(N,N-dimethylsulfamoyl)-6-ethoxy-8- methoxy-6H-isoindolo[2,l-a]indole-3-carboxamide was dissolved in THF (75 mL). To the solution was added a solution of 2 N HCl (300 mL). The mixture was vigorously stirred under N2 at rt for 16 h. The resulting suspension was filtered and washed with cooled TBME (2 X 30 mL). the filer cake was vacuum air dried overnight to give the title compound as a yellow solid. HPLC purity, 99% 1H NMR (DMSO-d6, 300 MHz) δ 11.65 (s, IH), 8.16 (s, IH), 7.76 (d, J= 5.9 Hz, IH), 7.73 (d, J= 5.9 Hz, IH), 7.58 (dd, J= 8.5 and 1.5 Hz, IH), 7.17-7.20 (m, 2H), 7.08 (dd, J = 8.5 and 1.4 Hz, IH), 6.55 (d, J= 8.6 Hz, IH), 3.86 (s, 3H), 3.14-3.18 (m, IH), 2.91 (s, 6H), 1.75-1.99 (m, 7H), 1.48-1.60 (m, 3H); m/z 484 (M + H)+.
Intermediate 7

7H-Indolo[2, 1-a] ‘ [2] benzazepine-6-carboxylic acid, 13-cyclohexyl-10- [[[(dimethylamino)sulfonyl] amino] carbonyl]-3-methoxy-, methyl ester. A mixture of the 3-cyclohexyl-N-(N,N-dimethylsulfamoyl)-2-(2-formyl-4-methoxyphenyl)-lH- indole-6-carboxamide (4.8g, 0.01 mol), methyl 2-(dimethoxyphosphoryl)acrylate (9.7 g, 0.02 mol) and cesium carbonate (7.1g, 0.02 mol) in DMF (28mL) was stirred for 20 hr at an oil bath temperature of 55 ° C. The mixture was poured into ice-water and acidified with dilute HCl to precipitate the crude product. The solid was collected, dried and flash chromatographed on Siθ2 (11Og) using an ethyl acetate and methylene chloride (1: 10) solution containing 2% acetic acid. Homogeneous fractions were combined and evaporated to afford the title compound as a pale yellow solid (3.9g, 71 % yield). MS: 552 (M=H+).
An alternate procedure for the preparation of 7H-indolo[2,l- a] [2]benzazepine-6-carboxylic acid, 13-cyclohexyl-10- [[[(dimethylamino)sulfonyl]amino]carbonyl]-3-methoxy-, methyl ester is provided below. A solution of l l-cyclohexyl-N-[(dimethylamino)sulfonyl]-6-hydroxy-8- methoxy-6H-isoindolo[2,l-a]indole-3-carboxamide (cyclic hemiaminal) (63.0 g, 130 mmol), methyl 2-(dimethoxyphosphoryl)acrylate (60 g, 261 mmol), cesium carbonate (106 g, 326 mmol) in DMF (400 mL) was heated at 60 °C (bath temp) for 4.5h. Additional methyl 2-(dimethoxyphosphoryl)acrylate (15 g, 65 mmol) and cesium carbonate (21.2 g, 65 mmol) were added and the reaction was heated at 60 °C overnight then and cooled to rt. The stirring reaction mixture was diluted with H2O (1 L), slowly neutralized with IN aqueous HCl (800 mL), stirred 3h, and then the precipitates were collected by filtration. The solids were triturated with Et20 (800 mL) and dried to yield methyl 7H-indolo[2,l-a][2]benzazepine-6-carboxylic acid, 13- cyclohexyl-10-[[[(dimethylamino)sulfonyl]amino]carbonyl]-3-methoxy-, methyl ester (70.2 g, 127 mmol, 98%) as a yellow solid which was used without further purification. IHNMR (300 MHz, CDC13) δ 8.67 (s, IH), 8.09 (s, IH), 7.86 (d, J = 8.4 Hz, IH), 7.80 (s, IH), 7.50 (d, J = 8.4 Hz, IH), 7.42 (d, J = 8.8 Hz, IH), 7.08 (dd, J = 2.6, 8.8 Hz, IH), 6.98 (d, J = 2.6 Hz, IH), 5.75 – 5.51 (m, IH), 4.29 – 4.01 (m, IH), 3.89 (s, 3H), 3.82 (s, 3H), 3.05 (s, 6H), 2.87 – 2.73 (m, IH), 2.11 – 1.12 (m, 10H). LCMS: m/e 550 (M-H)-, ret time 3.21 min, column A, 4 minute gradient.
Example 1

Cycloprop[d]indolo[2,l-a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (+/-)-. TBTU (43.7 mg, 0.136mmol) and DIPEA (0.095 mL, 0.544 mmol) were added to a solution of (+/-) cycloprop[d]indolo[2,l-a][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (50 mg, 0.0906 mmol) in DMSO (2.0 mL). The reaction mixture was stirred at rt for 15 min. 3-Methyl-3,8-diaza-bicyclo[3.2. l]octane dihydrochloride {J & W PharmLab, LLC Morrisville, PA 19067-3620}. (27.1 mg, 0. 136 mmol) was then added and the reaction mixture was stirred at rt for 3 hr. It was then concentrated and the residue was purified by preparative reverse phase HPLC to give the final product as a yellow solid, (32 mg, 46% yield). MS m/z 660(MH+), Retention time: 2.445 min IH NMR (300 MHz, MeOD) δ ppm 0.20 (m, 0.23 H) 1.11 – 2.25 (m, 15.77 H) 2.58 (m, 0.23 H) 2.69 (m, 0.77 H) 2.75 – 3.11 (m, 10 H) 3.28 – 3.75 (m, 5 H) 3.91 (s, 2.31 H) 3.92 (s, 0.69 H) 4.15 – 4.37 (m, 1 H) 4.68 (m ,br, 1 H) 4.94 – 5.00 (m, 0.23 H) 5.16 (d, J=15.00 Hz, 0.77 H) 7.00 – 7.09 (m, 1 H) 7.18 (d, J=2.56 Hz, 0.23 H) 7.21 (d, J=2.56 Hz, 0.77 H) 7.33 (d, J=8.41 Hz, 0.77 H) 7.35 (d, J=8.42 Hz, 0.23 H) 7.57 (dd, J=8.42, 1.46 Hz, 0.77 H) 7.62 (dd, J=8.78, 1.46 Hz, 0.23 H) 7.91 (d, J=8.42 Hz, 0.77 H) 7.93 (d, J=8.42 Hz, 0.23 H) 8.00 (s, 0.77 H) 8.07 (s, 0.23 H).
Example 4

Cycloprop[d]indolo[2,l-a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonylj ‘- 1 , Ia, 2, 12b-tetrahydro-ll-methoxy-la-[(8-methyl-3, 8- diazabicyclo[3.2.1]oct-3-yl)carbonyl]-, (+/-)-. To a solution of (+/-) cycloprop[d]indolo[2,l-a][2]benzazepine-5-carboxamide, 8-cyclohexyl-la-(3,8- diazabicyclo[3.2.1]oct-3-ylcarbonyl)-N-[(dimethylamino)sulfonyl]-l,la,2,12b- tetrahydro-11-methoxy- (54 mg, 0.071 mmol) in methanol (3 mL), paraformaldehyde (6.4 mg, 0.213 mmol), ZnCl2 (29 mg, 0.213 mmol) and
Na(CN)BH3 (13.4 mg, 0.213 mmol) were added. The resultant mixture was heated at 60°C for 2hr, and then cooled to rt. The solid present was removed by filtration, and the filtrate was concentrated under vacuum and the residue purified by preparative reverse phase HPLC to give the title compound as a light yellow colored solid, (37 mg, 67% yield). MS ml 660(MH+), Retention time: 2.495 min. IH NMR (500 MHz, MeOD) δ ppm 0.21 (m, 0.3 H) 1.13 (m, 0.3 H) 1.18 – 2.22 (m, 15.4 H) 2.58 (m, 0.3 H) 2.68 (m, 0.7 H) 2.76 – 3.11 (m, 11 H) 3.32 – 3.37 (m, 1 H) 3.63 (d, J=15.56 Hz, 0.7 H) 3.82 – 4.32 (m, 7.3 H) 4.88 – 4.92 (m, 0.3 H) 5.08 (d, J=15.56 Hz, 0.7 H) 7.00 – 7.08 (m, 1 H) 7.18 (d, J=2.14 Hz, 0.3 H) 7.21 (d, J=2.14 Hz, 0.7 H) 7.32 (d, J=8.55 Hz, 0.7 H) 7.35 (d, J=8.55 Hz, 0.3H) 7.57 (d, J=7.93 Hz, 0.7 H) 7.62 (dd, J=8.39, 1.37 Hz, 0.3 H) 7.91 (d, J=8.55 Hz, 0.7 H) 7.93 – 7.99 (m, 1 H) 8.09 (s, 0.3 H).
Example 6

Cycloprop [d] indolo [2, 1 -a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (laR,12bS)-. To a solution of (-) cycloprop[d]indolo[2,l-a][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (204 mg, 0.37 mmol) in DMSO (8.0 mL), TBTU (178 mg, 0.555 mmol) and DIPEA (0.39 mL, 2.22 mmol) were added. The reaction mixture was stirred at rt for 15 min. Then 3- methyl-3,8-diaza-bicyclo[3.2.1]octane dihydrochloride (111 mg, 0. 555 mmol) was added and the reaction mixture was stirred at rt for 2 hr. It was then concentrated and the residue was purified by preparative reverse phase HPLC to give a yellow solid as final TFA salt. (265 mg, 92% yield). Average Specific Rotation: -53.56° Solvent, MeOH.; Wavelength 589 nm; 50 cm cell. MS m/z 660(MH+), Retention time: 3.035 min. 1H NMR (300 MHz, MeOD) δ ppm 0.20 (m, 0.23 H) 1.11 – 2.25 (m, 15.77 H) 2.58 (m, 0.23 H) 2.69 (m, 0.77 H) 2.75 – 3.11 (m, 10 H) 3.28 – 3.75 (m, 5 H) 3.91 (s, 2.31 H) 3.92 (s, 0.69 H) 4.15 – 4.37 (m, 1 H) 4.68 (m ,br, 1 H) 4.94 – 5.00 (m, 0.23 H) 5.16 (d, J=15.00 Hz, 0.77 H) 7.00 – 7.09 (m, 1 H) 7.18 (d, J=2.56 Hz, 0.23 H) 7.21 (d, J=2.56 Hz, 0.77 H) 7.33 (d, J=8.41 Hz, 0.77 H) 7.35 (d, J=8.42 Hz, 0.23 H) 7.57 (dd, J=8.42, 1.46 Hz, 0.77 H) 7.62 (dd, J=8.78, 1.46 Hz, 0.23 H) 7.91 (d, J=8.42 Hz, 0.77 H) 7.93 (d, J=8.42 Hz, 0.23 H) 8.00 (s, 0.77 H) 8.07 (s, 0.23 H). An alternate procedure for the synthesis of cycloprop[d]indolo[2,l- a][2]benzazepine-5-carboxamide, 8-cyclohexyl-N-[(dimethylamino)sulfonyl]- l,la,2,12b-tetrahydro-l l-methoxy-la-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8- yl)carbonyl]-, (laR,12bS)-rel-(-)-is provided below. To a mixture of (-) cycloprop[<i]indolo[2,l-α][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (25.2 g, 45.68 mmol) and 3-methyl-3,8-diazabicyclo-[3.2.1]octane dihydrochloride (10.0 g, 50.22 mmol) in anhydrous MeCN (300 mL) was added DIPEA (23.62 g, 182.72 mmol) under N2. After 15 min, TBTU (16.12 g, 50.22 mmol) was added. The reaction solution was stirred for 30 min under N2. The ΗPLC indicated the disappearance of starting material. The solvent in the solution was evaporated to give a foam. This was dissolved in EtOAc (2.5 L), washed with H2O (1.5 L), H2O/brine (8:2) (1.5 L), brine (1.5 L), dried over Na2SO4 and evaporated to give 28.8 g of crude product. This solid was pooled with 45.4 g of material obtained from five separated reactions to afford a total of 74.2 g of crude product. This was passed through a pad of silica gel (E. Merck 230-400 mesh, 1 kg), eluting with MeOH/CH2Cl2 (2.5:97.5). After evaporation, it gave a foam, which was treated with EtOAc and hexane to turn into a solid. After drying at 50 °C under vacuum for 7 h, the GC analysis indicated it has 1.4% each of EtOAc and hexane. After further drying at 61-64 °C, the GC analysis indicated it still has 1.0% of hexane and 1.4% of EtOAc. The product was dissolved in Et2O and slowly evaporated in vacuum three times, dried at 60 °C under vacuum for 3 h to give 68.3 g. This was washed with H2O (900 mL) and redried at 68 °C under vacuum for 7 h to give 67.1 g (77% yield) of the compound of example 6. The GC analysis indicated it has 0.97% Of Et2O. HPLC conditions column: Cadenza CD-C18 3 x 250 mm; UV: 257 and 220 nm; 25 °C; flow rate: 0.5 mL/min; gradient time: 38 min, 0 – 80% B (0 – 35 min) and 80% B (35 – 38 min); solvent A: 25 nM CH3COONH4 at pH 4.7 in water, solvent B: MeCN. HPLC purity 99.7% (Rt 26.54 min); Chiral HPLC conditions column: Regis (S5S) Whelk-Ol 250 x 4.6 mm; UV 258nm; 35 °C; flow rate 2.0 mL/min; mobile phase C02/Me0H; gradient time 20 min, 30% MeOH (0 – 1 min), 30 – 48% MeOH (1 – 19 min), 48% MeOH (19 – 20 min). Chiral HPLC purity > 99.8% (Rt 16.60 min); LC/MS (ES+) 660.36 (M+H, 100); HRMS: calcd. 660.3220, found 660.3197; [α]D 25 C – 79.66 ° (c 1.06, MeOH); Anal. Calcd for C36H45N5O5S-O-O H2O»0.09 Et2O: C, 64.53; H, 7.00; N, 10.35; S, 4.74; H2O, 1.51; Et2O, 0.97. Found: C, 64.50; H, 7.12; N, 10.41; S, 5.14; H2O, 1.52; Et2O, 0.97. The absolute stereochemistry of cycloprop[d]indolo[2,l- a][2]benzazepine-5-carboxamide, 8-cyclohexyl-N-[(dimethylamino)sulfonyl]- l,la,2,12b-tetrahydro-l l-methoxy-la-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8- yl)carbonyl]-, (laR,12bS)-rel-(-)- is as drawn above, and was determined from an x- ray crystal structure obtained on the (R)-camphorsulfonic acid salt.
Additionally, the following salts were prepared: hydrochloride, phosphate, acetate, sulfate, camsylate, sodium, calcium, and magnesium. The hydrochloride salt had the following characteristics. DSC: small, broad endotherm from 25°C to 75°C, and potential melt/degradation endotherm with peak at temperatures ranging between 253 °C and 258 °C; TGA: Early weight loss from 25°C to 75°C ranging between 0.003% and 1.5%, and degradation weight loss starting at approximately 200°C.
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
| WO2006020082A1 * | Jul 15, 2005 | Feb 23, 2006 | Squibb Bristol Myers Co | Inhibitors of hcv replication |
| WO2006046030A2 * | Oct 25, 2005 | May 4, 2006 | Angeletti P Ist Richerche Bio | Tetracyclic indole derivatives as antiviral agents |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008111978A1 | Mar 13, 2007 | Sep 18, 2008 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2008112473A1 * | Mar 5, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112841A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112848A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112851A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv inhibitors |
| WO2008112863A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2009067108A1 * | Nov 20, 2007 | May 28, 2009 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2009067392A1 * | Nov 17, 2008 | May 28, 2009 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine derivatives for the treatment of hepatitis c |
| WO2009067481A1 * | Nov 19, 2008 | May 28, 2009 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2010080874A1 | Jan 7, 2010 | Jul 15, 2010 | Scynexis, Inc. | Cyclosporine derivative for use in the treatment of hcv and hiv infection |
| WO2013059265A1 * | Oct 17, 2012 | Apr 25, 2013 | Bristol-Myers Squibb Company | A compound for the treatment of hepatitis c |
| WO2014014885A1 * | Jul 16, 2013 | Jan 23, 2014 | Bristol-Myers Squibb Company | Novel methods and intermediates for the preparation of (4bs,5ar)-12-cyclohexyl-n-(n,n-dimethylsulfamoyl)-3-methoxy-5a-((1 r,5s) -3-methyl-3,8-diazabicyclo[3.2.1]octane-8-carbonyl)-4b,5,5a,6-tetrahydrobenzo [3,4]cyclopropa[5,6]azepino[1,2-a]indole-9-carboxamide |
| CN101679442B | Mar 13, 2008 | Feb 20, 2013 | 百时美施贵宝公司 | Compounds for the treatment of hepatitis c |
| EP2518073A1 * | Nov 19, 2008 | Oct 31, 2012 | Bristol-Myers Squibb Company | Compounds for the treatment of Hepatitis C |
The First Kilogram Synthesis of Beclabuvir, an HCV NS5B Polymerase Inhibitor
 , Kenneth J. Fraunhoffer, Zhinong Gao, Chao Hang, Yi Hsiao, Wenhao Hu, Kishta Katipally, Adam Littke, Aghogho Pedro, Yuping Qiu, Maria Sandoval, Richard Schild, Michelle Soltani, Anthony Tedesco, Dale Vanyo, Purushotham Vemishetti, and Robert E. Waltermire
, Kenneth J. Fraunhoffer, Zhinong Gao, Chao Hang, Yi Hsiao, Wenhao Hu, Kishta Katipally, Adam Littke, Aghogho Pedro, Yuping Qiu, Maria Sandoval, Richard Schild, Michelle Soltani, Anthony Tedesco, Dale Vanyo, Purushotham Vemishetti, and Robert E. Waltermire
The process development and kilogram-scale synthesis of beclabuvir (BMS-791325, 1) is described. The convergent synthesis features the use of asymmetric catalysis to generate a chiral cyclopropane fragment and coupling with an indole fragment via an alkylation. Subsequent palladium-catalyzed intramolecular direct arylation efficiently builds the central seven-membered ring. The target was prepared in 12 linear steps with five isolations in an overall yield of 8%.
Preparation of (4bS,5aR)-12-Cyclohexyl-N-(N,N-dimethylsulfamoyl)-3-methoxy-5a-((1R,5S)-3-methyl-3,8-diazabicyclo[3.2.1]octane-8-carbonyl)-4b,5,5a,6-tetrahydrobenzo[3,4]cyclopropa[5,6]azepino[1,2-a]indole-9-carboxamide Hydrochloride (1·HCl)
BMS-791325·HCl (1·HCl) was isolated in 89.5% yield.
1H NMR (600 MHz, 10:1 v/v CD3CN/D2O): major rotamer: 7.91 (br s, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.55 (br d, J = 8.5 Hz, 1H), 7.29 (d, J = 8.5 Hz, 1H), 7.20 (d, J = 2.5 Hz, 1H), 7.00 (dd, J = 8.5 Hz, 2.7 Hz, 1H), 5.03 (br d, J = 12.7 Hz, 1H), 4.58 (br d, J = 4.9 Hz, 2H), 3.87 (s, 3H), 3.56 (d, J = 15.5 Hz, 1H), 3.40 (br s, 3H), 3.32–3.28 (m, 4H), 2.96 (s, 6H), 2.92 (tt, J= 12.2, 3.6 Hz, 1H), 2.59 (br t, J = 7.0 Hz, 1H), 2.05–1.90 (m, 2H), 1.79–1.71 (m, 4H), 1.55 (br d, J= 12.2 Hz, 2H), 1.46–1.36 (m, 4H), 1.26 (t, J = 5.3 Hz, 2H), 1.23–1.15 (m, 2H);
minor rotamer: 8.05 (br s, 1H), 7.92 (d, J = 8.5 Hz, 1H), 7.58 (dd, J = 8.5, 1.4 Hz, 1H), 7.34 (d, J = 8.5 Hz, 1H), 7.15 (d, J = 2.6 Hz, 1H), 6.98 (d, 1H, overlap with major rotamer), 4.91 (d, J = 15.0 Hz, 1H), 4.58 (br d, J = 4.9 Hz, 2H), 4.11 (d, J = 15.0 Hz, 1H), 3.89 (s, 3H), 3.46 (br d, J = 12.5 Hz, 2H), 3.17 (br d, J = 12.5 Hz, 2H), 2.97 (s, 6H), 2.85 (br s, 3H), 2.76 (tt, J = 12.1, 3.5 Hz, 1H), 2.49 (br s, 1H), 2.05–1.90 (m, 2H), 1.79–1.71 (m, 4H), 1.46–1.36 (m, 6H), 1.23–1.15 (m, 2H), 1.10 (m, 1H), 0.03 (t, J = 6.1 Hz, 1H).
13C NMR (125 MHz, 10:1 v/v CD3CN/D2O): major rotamer: 170.1, 167.7, 161.0, 140.4, 139.3, 135.9, 133.6, 131.1, 124.9, 123.0, 121.7, 120.8, 119.0, 118.6, 114.3, 110.7, 59.2, 56.2, 53.1, 48.3, 44.5, 38.9, 37.6, 34.8, 33.77, 33.72, 27.92, 27.77, 26.82, 26.5, 23.6, 18.5;
minor rotamer: 168.3, 168.0, 161.3, 138.4, 137.5, 135.8, 134.2, 130.0, 125.4, 121.9, 120.0, 119.64, 119.58, 117.9, 113.3, 111.3, 59.6, 56.3, 53.1, 44.6, 42.2, 38.9, 38.3, 37.4, 33.8, 33.6, 28.3, 27.74, 26.79, 26.5, 24.84, 11.9.
HRMS (ESI) calcd for C36H45N5O5S (free base) [M + H]+660.3214, found m/z 660.3220.
////////