















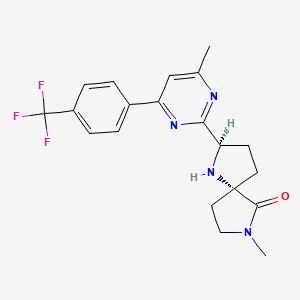
BIIB-095
ROTATION (+)
1493790-64-9 CAS free form,
1493772-48-7 cas Hcl salt
cas 1493790-65-0, 1496563-32-6 ,SULPHATE ???
cas 1496563-31-5 SULFATE 1;1
cas 1496563-32-6 SULFATE HYDRATE 1;1;1
(2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one
1,7-Diazaspiro[4.4]nonan-6-one, 7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)phenyl]-2-pyrimidinyl]-, (2R,5S)-
C20 H21 F3 N4 O, 390.40
- Originator Biogen
- Class Analgesics
- Mechanism of Action Nav1.7 voltage-gated sodium channel inhibitors
- Phase I Neuropathic pain
- 29 Mar 2018 Phase-I clinical trials in Neuropathic pain (In volunteers) in United Kingdom (PO) (NCT03454126)
- 05 Mar 2018 Biogen plans a phase I trial for Pain, including Neuropathic pain (In volunteers) in USA (PO) (NCT03454126)
- 05 Mar 2018 Preclinical trials in Neuropathic Pain in USA (PO), before March 2018
In March 2018, a randomized, double blind, placebo controlled, single and multiple-ascending dose, dose-escalation phase I study ( NCT03454126; 255HV101; 2017-003982-90) was initiated in the UK in healthy subjects (expected n = 80) to evaluate the safety, tolerability and pharmacokinetics of BIIB-095. At that time, the trial was expected to complete in December 2018
Biogen is developing BIIB-095, a voltage-gated sodium channel 1.7 inhibitor, for the potential oral treatment of neuropathic pain [2027279], [2027426]. In March 2018, a phase I trial was initiated in healthy subjects
Biogen is developing oral agent BIIB-095 for the treatment of chronic pain, including neuropathic pain. A phase I clinical trial is under way in healthy volunteers.
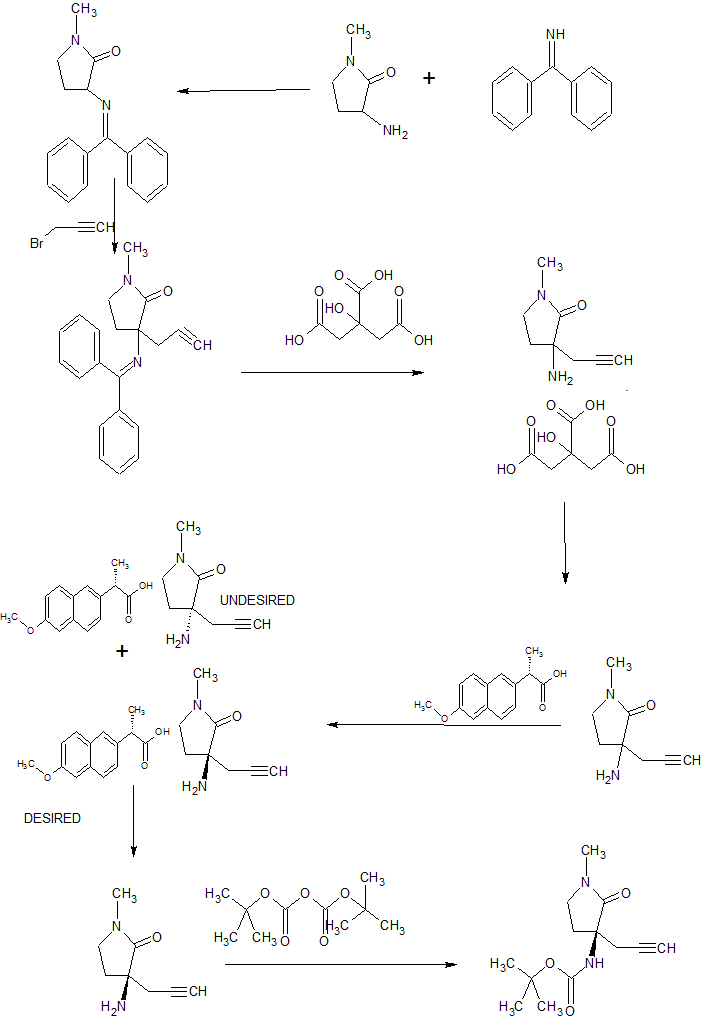
The compound was first claimed in WO2013175205 , for treating schizophrenia, assigned to subsidiary Convergence Pharmaceuticals Limited , naming some of the inventors. This might present the structure of BIIB-095 , a voltage-gated sodium channel 1.7 inhibitor, being developed by Biogen for the oral treatment of neuropathic pain; in March 2018, a phase I trial was initiated in healthy subjects.
PATENT
WO2013175205
CONTD………………
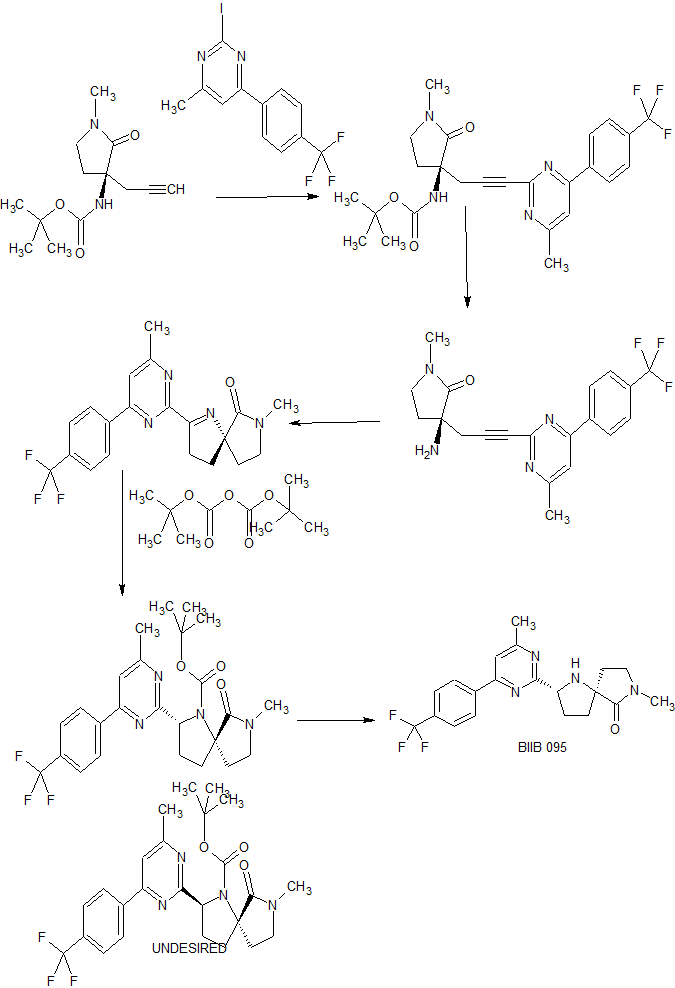
INTERMEDIATE
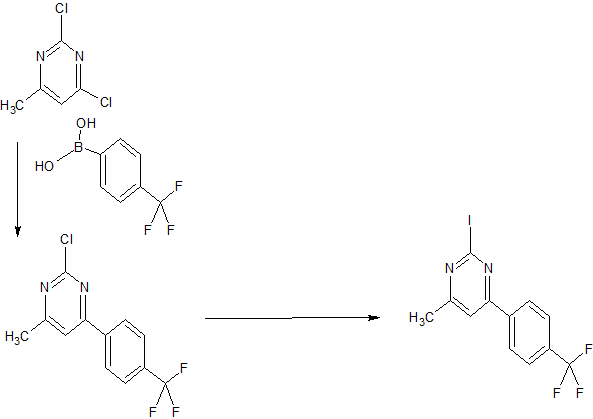
WO 2013175206
US 20150119404
https://patents.google.com/patent/US20150119404
Patent
WO-2019067961
Novel salts (citrate, mesylate, hydrosulfate, saccharinate and oxalate) forms of 7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-one, processes for their preparation and compositions comprising them are claimed. Also claimed are their use for treating diseases and conditions mediated by modulation of voltage-gated sodium channels.
Voltage-gated sodium channels are responsible for the initial phase of the action potential, which is a wave of electrical depolarisation usually initiated at the soma of the neuron and propagated along the axon to the terminals. At the terminals, the action potential triggers the influx of calcium and the release of neurotransmitter. Drugs, such as lidocaine, that block voltage-gated sodium channels are used as local anaesthetics. Other sodium channel blockers, such as lamotrigine and carbamazepine are used to treat epilepsy. In the latter case, partial inhibition of voltage-gated sodium channels reduces neuronal excitability and reduces seizure propagation. In the case of local anaesthetics, regional block of sodium channels on sensory neurons prevents the conduction of painful stimuli. A key feature of these drugs is their state-dependent mechanism of action. The drugs are thought to stabilise an inactivated conformation of the channel that is adopted rapidly after the channel opens. This inactivated state provides a refractory period before the channel returns to its resting (closed) state ready to be reactivated. As a result, state-dependent sodium channel blockers inhibit the firing of neurons at high frequency, for example in response to painful stimuli, and will help to prevent repetitive firing during periods of prolonged neuronal depolarisation that might occur, for example, during a seizure. Action potentials triggered at lower frequencies, for example in the heart, will not be significantly affected by these drugs, although the safety margin differs in each case, since at high enough concentrations each of these drugs is capable of blocking the resting or open states of the channels.
The voltage-gated sodium channel family is made up of 9 subtypes, four of which are found in the brain, NaV1.1 , 1.2, 1.3 and 1.6. Of the other subtypes, NaV1.4 is found only in skeletal muscle, NaV1.5 is specific to cardiac muscle, and NaV1.7, 1.8, and 1.9 are found
predominantly in sensory neurons. The hypothesised binding site for state-dependent sodium channel blockers is the local anaesthetic (LA) binding site in the inner vestibule of the pore on transmembrane S6 of domain IV. Critical residues are located in a highly conserved region among the different subtypes, thus presenting a challenge for the design of new subtype selective drugs. Drugs such as lidocaine, lamotrigine and carbamazepine do not distinguish between the subtypes. However, selectivity can be achieved, and can be further enhanced functionally, as a result of the different frequencies at which the channels operate.
Drugs that block voltage-gated sodium channels in a state-dependent manner are also used in the treatment of bipolar disorder, either to reduce symptoms of mania or depression, or as mood stabilisers to prevent the emergence of mood episodes. Clinical and preclinical evidence also suggests that state-dependent sodium channel blockers may help to reduce the symptoms of schizophrenia. For example, lamotrigine has been shown to reduce symptoms of psychosis induced by ketamine in healthy human volunteers, and furthermore, studies in patients suggest that the drug can augment the antipsychotic efficacy of some atypical antipsychotic drugs, such as clozapine or olanzapine. It is hypothesised that efficacy in these psychiatric disorders may result in part from a reduction of excessive glutamate release. The reduction in glutamate release is thought to be a consequence of sodium channel inhibition in key brain areas, such as the frontal cortex. However, interaction with voltage-gated calcium channels may also contribute to the efficacy of these drugs.
WO 2013/175205 (Convergence Pharmaceuticals Limited) describes (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one hydrochloride, sulfuric acid salt and sulfuric acid salt hydrate which are claimed to be modulators of voltage-gated sodium channels. The object of the invention is to identify alternative salts of said compound which have advantageous properties.
Example 1
(2R,5S)-7-Methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-
To a solution of (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one (which may be prepared in accordance with the procedure described in Example 1 of WO 2013/175205) (4.45g, 0.0114 mol) dissolved in absolute ethanol (66.82 ml, 15 vol) at 45 °C was added a solution of citric acid in ethanol (1 M, 1.05 equiv. 12 ml) over a period of 2-3 minutes. The solution was aged at 45 °C for a period of 1 hour. After 30 minutes a seed of citrate salt (0.1 wt%) was added and the mixture allowed to cool over approximately 2 hours and mature for 18 hours at ambient temperature (approximately 10-15 °C). Following maturation the salt was noted to be a very thick suspension (white) that required mobilisation with 20 ml additional ethanol and a further maturation period of 2 hours at ambient temperature. Filtration was carried out under vacuum and the vessel and cake rinsed with 15 ml ethanol. The de-liquored cake was dried further in a vacuum oven at 50 °C to provide 6.0 g of crystalline white solid (91 % yield).
H NMR (400MHz, DMSO-D6): δΗ 1.90-2.05 (2H, m), 2.10-2.20 (2H, m,), 2.20-2.30 (1 H, m), -2.50 (1 H, m, partially masked by solvent)), 2.55-2.68 (4H, m), 2.56 (3H, s), 2.79 (3H, s),
3.28-3.40 (2H, m), 4.79 (1 H, t, J= 8.0 Hz), 7.92 (2H, d, J = 8.4 Hz), 8.03 (1 H, s), 8.45 (2H, d, J= 8.8Hz) ppm, (exchangeables not reported)
Characterisation of Example 1
The XRPD of Example 1 is presented in FIG. 1 and the DSC/TGA of Example 1 is presented in FIG. 2. The citrate salt of Example 1 displayed the following characteristics:
1 endotherm onset: 171.82°C
peak maximum: 174.55°C
There was an endotherm post the main endotherm.
There was no weight reduction until ca 168°C had been reached. The weight reduction commenced with the start of the main endotherm and coincided with the endotherm post the main endotherm which indicated that this thermal event was the onset of compound decomposition and loss of citric acid. Thermal events >220°C were due to compound decomposition.
The XPRD data in FIG. 1 demonstrated that under different extremes of humidity indicate a stable crystalline form of the citrate salt of Example 1 with no tendency to form hydrates. This is supported by DSC/TGA data in FIG. 2 which show clear transitions and no evidence of solvates.
Aqueous solubility of the citrate salt (Example 1) = 22mg/ml (25°C).
Example 2
(2R,5S)-7-Methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-one ) salt (E2)
To a solution of (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one (which may be prepared in accordance with the procedure described in Example 1 of WO 2013/175205) (4.45g, 0.0114 mol) dissolved in absolute ethanol (66.82 ml, 15 vol) at 45 °C was added a solution of methanesulfonic acid in ethanol (1 M, 1.05 equiv. 12 ml) over a period of 2-3 minutes. The solution was aged at 45 °C for a period of 1 hour. After 10 minutes nucleation and gradual crystallisation was noted to afford a thick mixture. Additional ethanol was added (10 ml) to mobilise the suspension that was then allowed to cool over approximately 2 hours and mature for 18 hours at ambient temperature (approximately 10-15 °C). Following maturation the salt was noted to be a thin, mobile suspension (white) that was filtered under vacuum and the vessel and cake rinsed with 15 ml ethanol. The de-liquored cake was dried further in a vacuum oven at 50 °C to provide 4.0 g of crystalline white solid (72% yield).
H NMR (400MHz, DMSO-D6): δΗ 2.1-2.45 (4H, m), 2.27 (3H, s), 2.50-2.75 (2H, m), 2.61 (3H, s), 2.86 (3H, s), 3.35-3.50 (2H, m), 5.20 (1 H, t, J = 8 Hz), 7.96 (2H, d, J = 8.8 Hz), 8.17 (1 H, s), 8.51 (2H, d, J = 8.4Hz), 9.45 (1 H, br), 10.16 (1 H, br) ppm.
Characterisation of Example 2
The XRPD of Example 2 is presented in FIG. 3 and the DSC/TGA of Example 2 is presented in FIG. 4. The DSC thermograph of the methanesulfonate (mesylate) (Example 2) displayed the following characteristics:
One distinct endotherm onset: 247.34°C
peak maximum: 250.34°C
The TGA thermograph showed no weight reduction until ca 250°C had been reached. The weight reduction commenced with the start of the main endotherm and indicated that this thermal event was the onset of compound decomposition. There is no evidence of entrapped solvents or water.
The XPRD data in FIG. 3 demonstrated that under different extremes of humidity indicate a stable crystalline form of the mesylate salt of Example 2 with no tendency to form hydrates. This is supported by DSC/TGA data in FIG. 4 which show clear transitions and no evidence of solvates.
Aqueous solubility of the mesylate salt (Example 2) = 65mg/ml (25°C).
Example 3
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one hydrosulfate single crystals: 25.0 mg of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluorome
one hydrosulfate was added to 4 mL vial. 1.000 mL of anhydrous EtOH was added, and the sample was filtered. Anhydrous hexanes were added dropwise until the solution neared the precipitation point. The vial was sealed and left undisturbed for 24 hr, after which time a crop of single crystals was evident. The sample was sent for single crystal analysis and confirmed as the anhydrous (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate form (FIGs. 5A-5B).
Example 4
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one freebase: 8.00 g of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate (JM Lot R-2017-4323 D 301) was added to a 1 L Erlenmeyer flask and suspended and stirred vigorously in 400 mL of THF. 20% K2C03 (250 mL) was added and dissolved. The mixture was transferred to 1 L sep. funnel. 100 mL EtOAc was added and the aqueous and organic layers were separated. The aqueous layer was re-extracted with 50 mL of EtOAc and the combined organics were back-extracted with brine (100 mL) and water (100 mL). Due to fairly poor separation, a significant quantity of MgSCU was required to dry the solution. The solution was reduced via Rotavap (45 °C) to -50 mL, transferred to a 100 mL RB flask, reduced down to -10 mL, transferred to 20 mL scintillation vial and continued to be reduced to a thick oil. The oil was left on the Rotavap for another hour and a “wet” solid was obtained. Loosened solids on the bottom of the vial were left on the Rotavap for 1 hr with no heat applied to obtain a chunky solid. The contents was transferred to a mortar and pestle, ground to powder and fine granules, placed back in a 20 mL scintillation vial and left on a Rotavap overnight to obtain a dry solid (5.1 g). The XRPD pattern of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase is shown in FIG. 6.
Example 5
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one saccharinate: 199.7 mg of (2R,5S)-7-Methyl-2-(4-
methyl-6-(4-(trifluoromethyl)phenyl)pyrirnidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one free base (0.5115 mmol) was dissolved in 4.2 mL of 2-Me-THF. 98.1 mg of saccharin (0.5106 mmol) was dissolved in 4.2 mL of 2-Me-THF. Saccharin was added to the freebase, and after 15 seconds the mixture began to precipitate and solidify. 10 mL of 2-Me-THF was added and stirred at max rpm as to provide a thick white suspension in 10 min. The suspension was filtered, air dried under vacuum for 10 min on frit, then dried under a stream of nitrogen for 30 min resulting in 215 mg of white solid product. The XRPD pattern for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one saccharinate is shown in FIG. 7.
Example 6
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one oxalate: 403 mg of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase was dissolved in 4.03 mL EtOH. 1.000 mL of this solution was added to a 4 mL vial. 23.8 mg of oxalic acid was dissolved in 1.000 mL of EtOH and added dropwise to the stirring (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase solution. After 5 min, a white precipitate was evident and 2.000 mL of EtOH was added to the slurry to aid stirring. The resulting suspension was stirred overnight. The following day the suspension was filtered and dried on a frit under vacuum for 10 min yielding 106 mg of white solid. The XRPD pattern for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one oxalate is shown in FIG. 8.
Example 7
The single crystal structural information and refinement parameters for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate are shown in Table 1.
Table 1.
Largest peak, hole / e A-3 0.363, -0.264
The most prominent XRPD diffraction peaks were (2Θ): 7.8±0.2°, 8.1±0.2°, 12.6±0.2°, 14.3±0.2°, 16.5±0.2°, 18.5±0.2°, 19.6±0.2°, 24.8±0.2° and 25.3±0.2°.
PATENTS
US2018360833NOVEL PYRIMIDINYL-DIAZOSPIRO COMPOUNDS2018-06-27
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017304303 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2017-07-11 | |
| US9737536 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2016-05-25 | 2016-09-15 |
| US2016184306 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2016-02-15 | 2016-06-30 |
| US9309254 | NOVEL COMPOUNDS | 2013-05-22 | 2015-04-30 |
| US9376445 | NOVEL COMPOUNDS | 2013-05-22 | 2015-06-18 |
////////////////BIIB-095, BIIB095, BIIB 095, PHASE 1
CC1=NC(=NC(=C1)C2=CC=C(C=C2)C(F)(F)F)C3CCC4(N3)CCN(C4=O)C