
















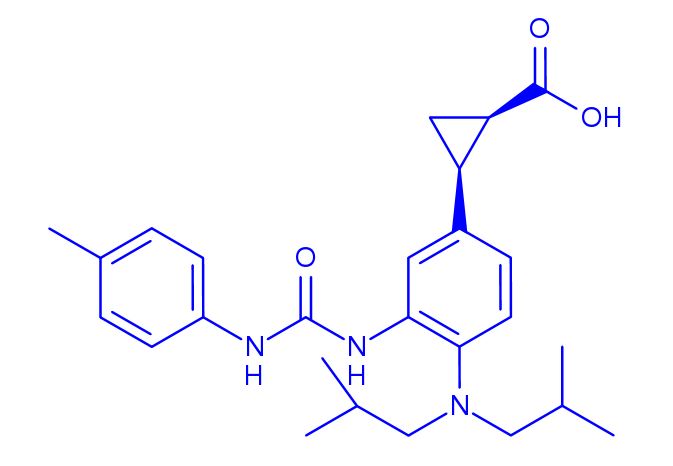

BMS-978587
| Molecular Formula: | C26H35N3O3 | CAS | 1629125-65-0 |
|---|---|---|---|
| Molecular Weight: | 437.582 |
US9675571 PATENT
Inventor James Aaron Balog Audris Huang Bin Chen Libing Chen Steven P. Seitz Amy C. Hart Jay A. Markwalder
AssigneeBristol-Myers Squibb Co Priority date 2013-03-15
IDO-IN-4; 1629125-65-0; SCHEMBL17456163; AKOS030526622; ZINC521836543; CS-5086
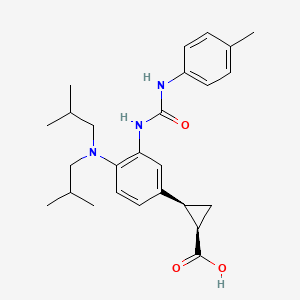
(1R,2S)-2-[4-(Di-isobutylamino)-3-(3-(p-tolyl)ureido)phenyl] Cyclopropanecarboxylic Acid
(1R,2S)-2-[4-[bis(2-methylpropyl)amino]-3-[(4-methylphenyl)carbamoylamino]phenyl]cyclopropane-1-carboxylic acid
(lR,2S)-2-(4-(diisobutylamino)-3-(3-(p- tolyl)ureido)phenyl)cyclopropanecarboxylic acid
BMS-978587 was discovered and developed within Bristol-Myers Squibb as a potent small molecule IDO inhibitor
Tryptophan is an amino acid which is essential for cell proliferation and survival. Indoleamine-2,3-dioxygenase is a heme-containing intracellular enzyme that catalyzes the first and rate-determining step in the degradation of the essential amino acid L-tryptophan to N-formyl-kynurenine. N-formyl-kynurenine is then metabolized by mutliple steps to eventually produce nicotinamide adenine dinucleotide (NAD+). Tryptophan catabolites produced from N-formyl-kynurenine, such as kynurenine, are known to be preferentially cytotoxic to T-cells. Thus an overexpression of IDO can lead to increased tolerance in the tumor microenvironment. IDO overexpression has been shown to be an independent prognostic factor for decreased survival in patients with melanoma, pancreatic, colorectal and endometrial cancers among others. Moreover, IDO has been found to be implicated in neurologic and psychiatric disorders including mood idsorders as well as other chronic diseases characterized by IDO activation and tryptophan depletiion, such as viral infections, for example AIDS, Alzheimer’s disease, cancers including T-cell leukemia and colon cancer, autimmune diseases, diseases of the eye such as cataracts, bacterial infections such as Lyme disease, and streptococcal infections.
Accordingly, an agent which is safe and effective in inhibiting production of IDO would be a most welcomed addition to the physician’s armamentarium
SYNTHESIS
PATENT
https://patents.google.com/patent/US9675571




Example 1 Method A Enantiomer 1 and Enantiomer 2 Enantiomer 1: (1R,2S)-2-(4-(diisobutylamino)-3-(3-(p-tolyl)ureido)phenyl)cyclopropanecarboxylic acid

PATENT
WO2014/150677
https://patents.google.com/patent/WO2014150677A1/en
Example 1- Method A
Enantiomer 1 and Enantiomer 2
Enantiomer 1 : (lR,2S)-2-(4-(diisobutylamino)-3-(3-(p- tolyl)ureido)phenyl)cyclopropanecarboxylic acid
Enantiomer 2: (lS,2R)-2-(4-(diisobutylamino)-3-(3-(p- tolyl)ureido)phenyl)cyclopropanecarboxylic acid

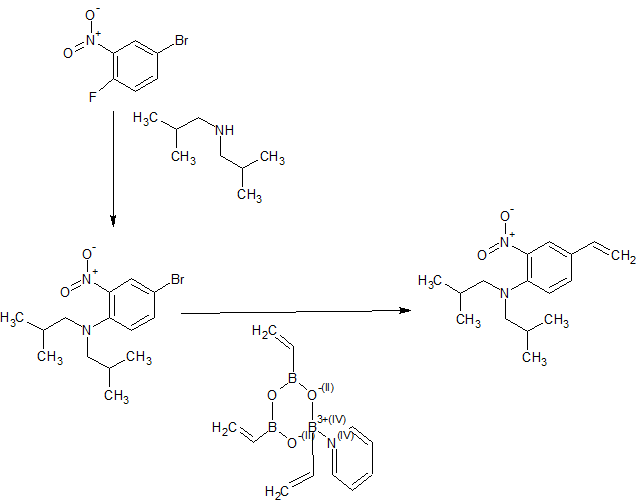
1A. 4-bromo-N,N-diisobutyl-2-nitroaniline
4-bromo-l-fluoro-2 -nitrobenzene (7 g, 31.8 mmol) and diisobutylamine (12.23 ml, 70.0 mmol) were heated at 130 °C for 3 h. It was then cooled to RT, purification via flash chromatography gave 1A (bright red solid, 8.19 g, 24.88 mmol, 78 % yield) LC-MS Anal. Calc’d for Ci4H2iBrN202 328.08, found [M+3] 331.03, Tr = 2.63 min (Method A).
IB. N,N-diisobutyl-2-nitro-4-vinylaniline
To a solution of 1 A (1 g, 3.04 mmol) in ethanol (15.00 mL) and toluene (5 mL) (sonication to break up the solid) was added 2,4,6-trivinyl- 1 ,3 ,5 ,2,4,6-trioxatriborinane pyridine complex (0.589 g, 3.64 mmol) followed by K3PO4 (1.289 g, 6.07 mmol) and water (2.000 mL). The reaction mixture was purged with Argon for 2 min and then Pd (PPh3)4(0.351 g, 0.304 mmol) was added. It was then heated at 80 °C in an oil bath for 8 h. LC-MS indicated completion. It was diluted with EtOAc (10 mL) and water (5 mL) and filtered through a pad of Celite, rinsed with EtOAc (2×30 mL). Aqueous layer was further extracted with EtOAc (2×30 mL), the combined extracts were washed with water, brine, dried over MgS04, filtered and concentrated. Purification via fiash chromatography gave IB (orange oil, 800 mg, 2.89 mmol, 95 % yield). LC-MS Anal. Calc’d for
Ci6H24N202 276.18, found [M+H] 277.34, Tr = 2.41 min (Method A). 1H NMR
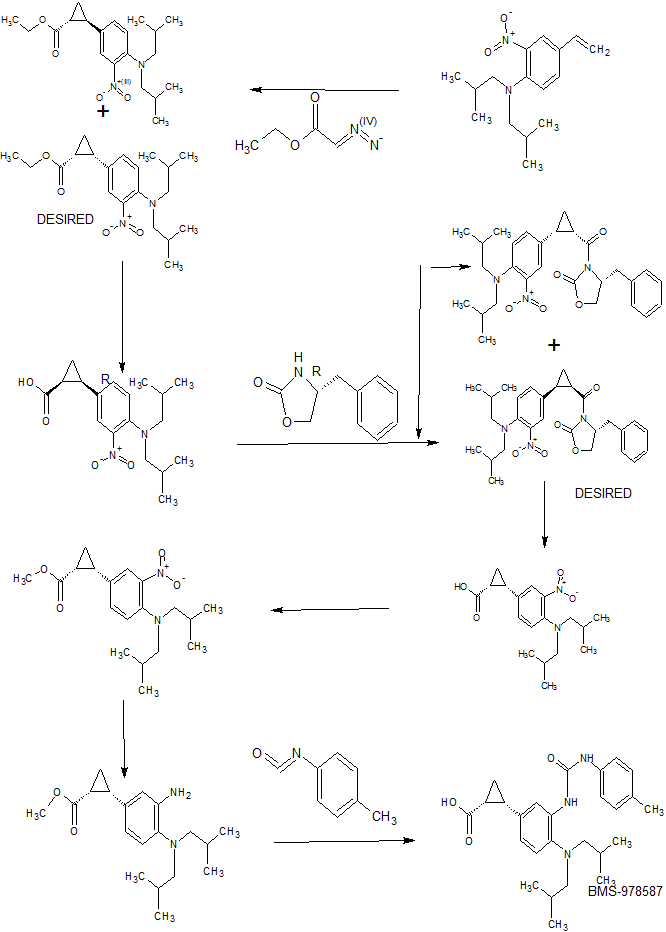
(400MHz, CHLOROFORM-d) δ 7.73 (d, J=2.2 Hz, 1H), 7.44 (dd, J=8.8, 2.2 Hz, 1H), 7.08 (d, J=8.6 Hz, 1H), 6.60 (dd, J=17.5, 10.9 Hz, 1H), 5.63 (dd, J=17.6, 0.4 Hz, 1H), 5.20 (d, J=11.2 Hz, 1H), 3.00 – 2.89 (m, 4H), 1.99 – 1.85 (m, 2H), 0.84 (d, J=6.6 Hz, 12H) IC. Racemic (lR,2S)-ethyl 2-(4-(diisobutylamino)-3 nitrophenyl)
cyclopropanecarboxylate
To a solution of IB (800 mg, 2.61 mmol) in DCM (15 mL) was added rhodium(II) acetate dimer (230 mg, 0.521 mmol) followed by a slow addition of a solution of ethyl diazoacetate (0.811 mL, 7.82 mmol) in CH2CI2 (5.00 mL) over a period of 2 h via a syringe pump. The reaction mixture turned into a dark red solution and it was stirred at RT for extra 1 h. LC-MS indicated the appearance of two peaks with the desired molecular mass, the solvent was removed in vacuo and purification via flash
chromatography gave 1C (cis isomer) (yellow oil, 220 mg, 0.607 mmol, 23.30 % yield) and trans isomer (yellow oil, 300 mg, 0.828 mmol, 31.8 % yield). LC-MS Anal. Calc’d for C20H30N2O4 362.22, found [M+H] 363.27, Tr = 2.34 min (cis), 2.42 min (trans) (Method A), cis isomer: 1H NMR (400MHz, CHLOROFORM-d) δ 7.62 (d, J=1.8 Hz, 1H), 7.30 – 7.25 (m, 1H), 7.02 (d, J=8.6 Hz, 1H), 3.95 – 3.86 (m, 2H), 2.89 (d, J=7.3 Hz, 4H), 2.53 – 2.44 (m, 1H), 2.07 (ddd, J=9.2, 7.9, 5.7 Hz, 1H), 1.87 (dquin, J=13.5, 6.8 Hz, 2H), 1.67 (dt, J=7.3, 5.5 Hz, 1H), 1.37 – 1.30 (m, 1H), 0.99 (t, J=7.0 Hz, 3H), 0.82 (d, J=6.6 Hz, 12H) trans isomer: 1H NMR (400MHz, CHLOROFORM-d) δ 7.43 (d, J=2.2 Hz, 1H), 7.17 – 7.11 (m, 1H), 7.08 – 7.03 (m, 1H), 4.18 (q, J=7.3 Hz, 2H), 2.89 (d, J=7.3 Hz, 4H), 2.46 (ddd, J=9.2, 6.4, 4.2 Hz, 1H), 1.94 – 1.80 (m, 3H), 1.62 – 1.54 (m, 1H), 1.34 – 1.23 (m, 4H), 0.83 (d, J=6.6 Hz, 12H)
ID. Racemic (lR,2S)-ethyl 2-(3-amino-4-(diisobutylamino)phenyl) cyclopropanecarboxylate
To a stirred solution of 1C (cis isomer) (220 mg, 0.607 mmol) in EtOAc (6 mL) was added palladium on carbon (64.6 mg, 0.061 mmol) and the suspension was hydrogenated (1 atm, balloon) at RT for 1 h. LC-MS indicated completion. The suspension was filtered through a pad of Celite and the filter cake was rinsed with EtOAc (2×30 mL). Combined filtrate and rinses were evaporated in vacuo. Purification via flash chromatography gave ID (light yellow oil, 140 mg, 0.421 mmol, 69.4 % yield). LC-MS Anal. Calc’d for C20H32N2O2 332.25, found [M+H] 333.34, Tr= 2.22 min (Method A). 1H NMR (400MHz, CHLOROFORM-d) δ 6.95 (d, J=8.1 Hz, 1H), 6.65 (d, J=2.0 Hz, 1H), 6.64 – 6.59 (m, 1H), 4.06 (s, 2H), 3.87 (qd, J=7.1, 0.9 Hz, 2H), 2.56 (d, J=7.0 Hz, 4H), 2.47 (q, J=8.6 Hz, IH), 2.01 (ddd, J=9.4, 7.8, 5.7 Hz, IH), 1.78 – 1.61 (m, 3H), 1.24 (ddd, J=8.6, 7.9, 5.1 Hz, IH), 0.92 (t, J=7.2 Hz, 3H), 0.89 (dd, J=6.6, 0.9 Hz, 12H)
Racemic example 1. Racemic (lR,2S)-2-(4-(diisobutylamino)-3-(3-(p- tolyl)ureido)phenyl)cyclopropanecarboxylic acid
To a solution of ID (140 mg, 0.421 mmol) in THF (4mL) was added 1- isocyanato-4-methylbenzene (0.079 mL, 0.632 mmol). The resulting solution was stirred at RT for 3 h. LC-MS indicated completion. The reaction mixture was concentrated and used without purification in the next step. The crude ester (180 mg, 0.387 mmol) was dissolved in THF (4 mL), NaOH (IN aqueous) (1.160 mL, 1.160 mmol) was added. Then MeOH (1 mL) was added to dissolve the precipitate and it turned into a clear yellow solution. After 60 h, reaction was complete by LC-MS. Most MeOH and THF was removed in vacuo and the crude was diluted with 2 mL of water, the pH was adjusted to ca. 2 using IN aqueous HC1. The aqueous phase was then extracted with EtOAc (3×10 mL) and the combined organic phase was washed with brine, dried over Na2S04 and concentrated. Purification via flash chromatography gave racemic example 1 (yellow foam, 110 mg, 0.251 mmol, 65.0 % yield), LC-MS Anal. Calc’d for CzeHssNsOs 437.27, found [M+H] 438.29, Tr = 4.22 min (Method A). 1H NMR (400MHz, CHLOROFORM- d) δ 10.15 (br. s., IH), 7.42 – 7.35 (m, 3H), 7.22 – 7.14 (m, 2H), 7.10 (d, J=8.1 Hz, 2H), 3.22 (d, J=6.6 Hz, 4H), 2.54 (q, J=8.6 Hz, IH), 2.31 (s, 3H), 2.16 – 1.98 (m, 3H), 1.61 (dt, J=7.3, 5.6 Hz, IH), 1.40 (td, J=8.3, 5.3 Hz, IH), 1.01 (br. s., 12H)
Example 1, Enantiomer 1 and Enantiomer 2. Chiral separation of racemic example 1 (Method H) gave enantiomer 1 Tr = 9.042 min (Method J). [a]24 D = -11.11 (c 7.02 mg/mL, MeOH) and enantiomer 2 Tr = 10.400 min (Method J). [a]24 D = + 11.17 (c 7.02 mg/mL, MeOH) as single enantiomers. Absolute stereochemistry was confirmed in example 1 method B.
Enantiomer 1 : LC-MS Anal. Calc’d for C26H35N3O3 437.27, found [M+H] 438.25, Tr= 4.19 min (Method A). 1H NMR (400MHz, CHLOROFORM-d) δ 8.12 (d, J=1.3 Hz, IH), 7.97 (s, IH), 7.20 (d, J=8.4 Hz, 2H), 7.14 – 7.07 (m, 2H), 7.02 (t, J=7.7 Hz, 2H),
6.89 (dd, J=8.1, 1.5 Hz, IH), 2.60 (q, J=8.6 Hz, IH), 2.50 (d, J=7.0 Hz, 4H), 2.32 (s, 3H), 2.13 – 2.04 (m, 1H), 1.71 – 1.55 (m, 3H), 1.35 (td, J=8.3, 5.1 Hz, 1H), 0.76 (dd, J=6.6, 2.2 Hz, 12H)
Enantiomer 2: LC-MS Anal. Calc’d for C26H35N3O3 437.27, found [M+H] 438.24, Tr= 4.18 min (Method A). 1H NMR (400MHz, CHLOROFORM-d) δ 8.11 (d, J=1.5 Hz, 1H), 7.96 (s, 1H), 7.23 – 7.16 (m, 2H), 7.13 – 7.07 (m, 2H), 7.05 – 6.98 (m, 2H), 6.89 (dd, J=8.3, 1.7 Hz, 1H), 2.59 (q, J=8.7 Hz, 1H), 2.49 (d, J=7.3 Hz, 4H), 2.32 (s, 3H), 2.12 – 2.03 (m, 1H), 1.70 – 1.53 (m, 3H), 1.34 (td, J=8.2, 5.0 Hz, 1H), 0.75 (dd, J=6.6, 2.0 Hz, 12H) Example 1 – Method B
Enantiomer 1 and Enantiomer 2
Enantiomer 2: (lS,2R)-2-(4-(diisobutylamino)-3-(3-(p- tolyl)ureido)phenyl)cyclopropanecarboxylic acid

IE. 4-(5,5-dimethyl-l,3,2-dioxaborinan-2-yl)-N,N-diisobutyl-2-nitroaniline
1A (10 g, 30.4 mmol), 5,5,5′,5′-tetramethyl-2,2′-bi(l,3,2-dioxaborinane) (7.55 g, 33.4 mmol), PdCl2(dppf)- CH2C12 adduct (0.556 g, 0.759 mmol) and potassium acetate
(8.94 g, 91 mmol) were combined in a round bottom flask, and DMSO (100 mL) was added. It was vacuated and back-filled with N2 three times, then heated at 80 °C for 8 h. Reaction was complete by LC-MS. Cooled to RT and passed through a short plug of silica gel, rinsed with a mixture of Hexane/EtOAc (5: 1) (3×100 mL). After removing the solvent in vacuo, purification via flash chromatography gave IE (orange oil, 9 g, 22.36 mmol, 73.6 % yield), LC-MS Anal. Calc’d for C19H31BN2O4 362.24, found [M+H] 295.18 (mass of boronic acid), Tr = 3.65 min (Method A). 1H NMR (400MHz,
CHLOROFORM-d) δ 8.13 (d, J=1.8 Hz, 1H), 7.73 (dd, J=8.4, 1.5 Hz, 1H), 7.04 (d, J=8.6 Hz, 1H), 3.75 (s, 4H), 3.00 – 2.92 (m, 4H), 1.93 (dquin, J=13.5, 6.8 Hz, 2H), 1.02 (s, 6H), 0.93 – 0.79 (m, 12H)
IF. (lS,2R)-ethyl 2-(4-(diisobutylamino)-3-nitrophenyl)
cyclopropanecarboxylate
To IE (9 g, 22.36 mmol) in a 500 mL round bottom flask was added 1,4-dioxane (60 mL). After it was dissolved, cesium carbonate (15.30 g, 47.0 mmol) was added. To the suspension was then added water (30 mL) slowly. It became an homogeneous solution. Enantiopure (lR,2R)-ethyl 2-iodocyclopropanecarboxylate (5.90 g, 24.59 mmol) (For synthesis see Organic Process Research & Development 2004, 8, 353-359 ) was then added. The resulting mixture was purged with nitrogen for 25 min. Then PdCl2(dppf)-
CH2C12 adduct (1.824 g, 2.236 mmol) was added. The reaction mixture was purged with nitrogen for another 10 min. It became dark brown colored solution. This mixture was then stirred under nitrogen at 87 °C for 22 h. LC-MS indicated product formation and depletion of starting material. It was then cooled to RT. After removing solvent under reduced pressure, it was diluted with EtOAc (50 mL) and water (50 mL). Organic layer was separated and the aqueous layer was further extracted with EtOAc (3x 30 mL). The combined organic layers were washed with brine, dried over MgS04, filtered and concentrated. Purification via flash chromatography gave IF (dark orange oil, 3.2 g, 8.83 mmol, 39.5 % yield), LC-MS Anal. Calc’d for C20H30N2O4 362.22, found [M+H] 363.3, Tr = 3.89 min (Method A). 1H NMR (400MHz, CHLOROFORM-d) 57.65 – 7.60 (m, 1H), 7.29 (d, J=2.2 Hz, 1H), 7.02 (d, J=8.6 Hz, 1H), 3.95 – 3.84 (m, 2H), 2.89 (d, J=7.3 Hz, 4H), 2.48 (q, J=8.6 Hz, 1H), 2.07 (ddd, J=9.2, 7.9, 5.7 Hz, 1H), 1.87 (dquin, J=13.5, 6.8 Hz, 2H), 1.67 (dt, J=7.3, 5.5 Hz, 1H), 1.38 – 1.28 (m, 1H), 0.99 (t, J=7.2 Hz, 3H), 0.82 (d, J=6.6 Hz, 12H
IG. (lS,2R)-ethyl 2-(3-amino-4-(diisobutylamino)phenyl)
cyclopropanecarboxylate
To a stirred solution of IF (5.5 g, 15.17 mmol) in EtOAc (150 mL) was added palladium on carbon (1.615 g, 1.517 mmol) and the suspension was hydrogenated (1 atm, balloon) for 1.5 h. LC-MS indicated completion. The suspension was filtered through a pad of Celite and the filter cake was rinsed with EtOAc (2×50 mL). Combined filtrate and rinses were concentrated under reduced pressure. Purification via flash chromatography gave 1G (yellow oil, 4.5 g, 13.53 mmol, 89 % yield). LC-MS Anal. Calc’d for
C20H32N2O2 332.25, found [M+H] 333.06, Tr = 2.88 min (Method A). 1H NMR
(400MHz, CHLOROFORM-d) δ 6.95 (d, J=7.9 Hz, 1H), 6.68 – 6.58 (m, 2H), 4.06 (s, 2H), 3.93 – 3.81 (m, 2H), 2.57 (d, J=7.3 Hz, 4H), 2.47 (q, J=8.6 Hz, 1H), 2.01 (ddd, J=9.4, 7.8, 5.5 Hz, 1H), 1.78 – 1.59 (m, 3H), 1.30 – 1.18 (m, 1H), 0.92 (t, J=7.2 Hz, 3H), 0.89 (dd, J=6.6, 0.9 Hz, 12H)
Example 1 enantiomer 2 was prepared following the reduction, urea formation and basic saponification procedures in racemic example 1 method A except that saponification was carried out at 50 °C for 8 h instead of at RT. Chiral analytical analysis verified it was enantiomer 2 Tr = 10.646 min (Method J). Absolute stereochemistry was confirmed by referring to reference: Organic Process Research & Development 2004, 8, 353-359.
Enantiomer 1 Method B: (lR,2S)-2-(4-(diisobutylamino)-3
tolyl)ureido)phenyl)cyclopropanecarboxylic acid

1H. Single enantiomer (lR,2S)-ethyl 2-(3-amino-4-(diisobutylamino)phenyl) cyclopropanecarboxylate
1H was prepared following procedures in example 1 enantiomer 2 method B utilizing enantiopure (l S,2S)-ethyl 2-iodocyclopropanecarboxylate. This was obtained through chiral resolution modifying the procedure in Organic Process Research & Development 2004, 8, 353-359, using (i?)-(+)-N-benzyl-a-methylbenzylamine instead of (S)-(-)-N-benzyl-a-methylbenzylamine). LC-MS Anal. Calc’d for C20H32N2O2 332.25, found [M+H] 333.06, Tr = 2.88 min (Method A). 1H NMR (400MHz, CHLOROFORM- d) δ 6.95 (d, J=7.9 Hz, 1H), 6.68 – 6.58 (m, 2H), 4.06 (s, 2H), 3.93 – 3.81 (m, 2H), 2.57 (d, J=7.3 Hz, 4H), 2.47 (q, J=8.6 Hz, 1H), 2.01 (ddd, J=9.4, 7.8, 5.5 Hz, 1H), 1.78 – 1.59 (m, 3H), 1.30 – 1.18 (m, 1H), 0.92 (t, J=7.2 Hz, 3H), 0.89 (dd, J=6.6, 0.9 Hz, 12H).
Note: 1H was also made through chiral separation (Method I) of racemic (1R,2S)- ethyl 2-(3-amino-4-(diisobutylamino)phenyl)cyclopropanecarboxylate. Chiral analytical analysis (Method K) showed 1H as a single enantiomer (99 % ee).
Example 1 enantiomer 1 was prepared following the reduction, urea formation and basic saponification procedures in racemic example 1 method A using 1H except that saponification was carried out at 50 °C for 8 h instead of at RT. Chiral analytical analysis verified it was enantiomer 1 with 97.8% ee (Method J).
Example 1 – Method C
Enantiomer 1
(lR,2S)-2-(4-(diisobutylamino)-3-(3-(p-tolyl)ureido)phenyl)cyclopropanecarboxylic acid

II. Diastereomer 1: (R)-4-benzyl-3-((lR,2S)-2-(4-(diisobutylamino)-3- nitrophenyl)cyclopropanecarbonyl)oxazolidin-2-one
Diastereomer 2: (R)-4-benzyl-3-((l S,2R)-2-(4-(diisobutylamino)-3- nitrophenyl)cyclopropanecarbonyl)oxazolidin-2-one: 1C (1.2 g, 3.31 mmol) was dissolved in THF (20 mL), NaOH (IN aqueous) (8.28 mL, 8.28 mmol) was added. Saw precipitate formed, then MeOH (5.00 mL) was added and it turned into a clear yellow solution. The reaction was monitored by LC-MS. After 24 h, reaction was complete. Most MeOH and THF was removed in vacuo and the crude was diluted with 10 mL of water, the pH was adjusted to ca. 2 using IN aqueous HC1. The aqueous phase was then extracted with EtOAc (3×30 mL) and the combined organic phase was washed with brine, dried over Na2S04 , filtered and concentrated to give 1.1 g of desired acid as an orange foam. This was used without purification in the subsequent step. To a solution of the crude acid from the previous step (1132 mg, 3.39 mmol) in THF (15 mL) cooled in an ice-water bath was added N-methylmorpholine (0.447 mL, 4.06 mmol) followed by slow addition of pivaloyl chloride (0.500 mL, 4.06 mmol). After stirring in an ice-water bath for 30 min, the reaction mixture was then cooled to -78 °C. In a separate reaction flask, ftBuLi (1.354 mL, 3.39 mmol) was added dropwise to a solution of (R)-4- benzyloxazolidin-2-one (600 mg, 3.39 mmol) in THF (15.00 mL). After 45 min at -78 °C, the solution was cannulated into the -78 °C anhydride mixture. After 30 min, the cooling bath was removed and the solution was allowed to warm to RT. After 1 h, LC-MS indicated completion. The reaction was quenched by addition of saturated aqueous NH4C1. The solution was then partitioned between EtOAc and water. The organic phase was further extracted with EtOAc (2×30 mL). The combined organic extracts were washed with water, brine, dried over MgS04, filtered and concentrated. Purification via flash chromatography gave II Diastereomer 1 (yellow oil, 600 mg, 1.216 mmol, 35.9 % yield). Diastereomer 2 (yellow oil, 450 mg, 0.912 mmol, 26.9 % yield) LC-MS Anal. Calc’d for C28H35N305 493.26, found: [M+H] 494.23, Tr = 5.26 min (Diastereomer 1). Tr = 5.25 min (Diastereomer 2) (Method A). Diastereomer 1 : 1H NMR (400MHz,
CHLOROFORM-d) δ 7.56 (d, J=1.8 Hz, 1H), 7.35 – 7.23 (m, 4H), 7.18 – 7.12 (m, 2H), 7.03 (d, J=8.8 Hz, 1H), 4.37 (ddt, J=9.6, 7.3, 3.6 Hz, 1H), 4.11 – 4.06 (m, 2H), 3.48 – 3.40 (m, 1H), 3.22 (dd, J=13.4, 3.5 Hz, 1H), 2.89 (d, J=7.3 Hz, 4H), 2.77 – 2.66 (m, 2H), 1.97 – 1.81 (m, 3H), 1.52 – 1.44 (m, 1H), 0.82 (d, J=6.6 Hz, 12H); Diastereomer 2: 1H NMR (400MHz, CHLOROFORM-d) δ 7.62 (d, J=2.0 Hz, 1H), 7.36 – 7.19 (m, 4H), 7.09 – 6.97 (m, 3H), 4.45 (ddt, J=10.2, 7.2, 3.0 Hz, 1H), 4.14 – 4.05 (m, 2H), 3.45 – 3.36 (m, 1H), 2.80 (d, J=7.3 Hz, 4H), 2.52 (dd, J=13.3, 3.2 Hz, 1H), 2.19 (dd, J=13.2, 10.3 Hz, 1H), 2.03 (dt, J=7.2, 5.8 Hz, 1H), 1.72 (dquin, J=13.4, 6.8 Hz, 2H), 1.45 (ddd, J=8.3, 7.3, 5.3 Hz, 1H), 0.64 (dd, J=6.6, 2.0 Hz, 12H) 1 J. (lR,2S)-methyl 2-(4-(diisobutylamino)-3-nitrophenyl)
cyclopropanecarboxylate
To a solution of II Diastereomer 1 (460 mg, 0.932 mmol) in THF (6mL) at 0 °C was added hydrogen peroxide (0.228 mL, 3.73 mmol). Then a solution of lithium hydroxide monohydrate (44.6 mg, 1.864 mmol) in water (2.000 mL) was added to the cold THF solution and stirred for 6 h. LC-MS indicated completion, then 2 mL of saturated aqueous Na2S03 was added followed by 3 mL of saturated aqueous NaHC03. The mixture was concentrated to remove most of the THF. The solution was then diluted with 5 mL of water. The aqueous solution was acidified with 1 N aqueous HC1 and extracted with EtOAc (3×20 mL). The combined organic extracts was washed with water, brine, dried over MgS04, filtered and concentrated to give 300 mg acid. To a solution of the crude acid from previous step (300 mg, 0.897 mmol) in MeOH (10 mL) was added 6 drops of concentrated H2SO4. The resulting solution was stirred at 50 °C for 6 h. After LC-MS indicated completion, solvent was removed under reduced pressure. It was then diluted with 5 mL of water, the aqueous layer was then extracted with EtOAc (3×20 mL) and the combined organic extracts were washed with water, brine, dried with Na2S04, filtered and concentrated. Purification via flash chromatography gave 1J (orange oil, 260 mg, 0.746 mmol, 83 % yield). LC-MS Anal. Calc’d for Ci9H28N204 348.20, found:
[M+H] 349.31 , Tr = 3.87 min (Method A). 1H NMR (400MHz, CHLOROFORM-d) δ
7.66 – 7.61 (m, 1H), 7.31 – 7.25 (m, 1H), 7.04 (d, J=8.8 Hz, 1H), 3.47 (s, 3H), 2.90 (d, J=7.3 Hz, 4H), 2.54 – 2.44 (m, 1H), 2.14 – 2.04 (m, 1H), 1.89 (dquin, J=13.5, 6.8 Hz, 2H),
1.67 (dt, J=7.5, 5.5 Hz, 1H), 1.42 – 1.31 (m, 1H), 0.83 (dd, J=6.6, 1.1 Hz, 12H)
IK. (lR,2S)-methyl 2-(3-amino-4-(diisobutylamino)phenyl)
cyclopropanecarboxylate
To a stirred solution of 1 J (100 mg, 0.287 mmol) in EtOAc (5mL) was added palladium on carbon (30.5 mg, 0.029 mmol) and the suspension was hydrogenated (1 atm, balloon) for 2 h. LC-MS indicated completion. The suspension was filtered through a pad of Celite and the filter cake was rinsed with EtOAc (20 mL). Combined filtrate and rinses were concentrated. Purification via flash chromatography gave IK (yellow oil, 90 mg, 0.287 mmol, 99 % yield). LC-MS Anal. Calc’d for Ci9H3oN202 318.23, found:
[M+H] 319.31 , Tr = 2.72 min (Method A). 1H NMR (400MHz, CHLOROFORM-d) δ 6.95 (d, J=8.1 Hz, 1H), 6.65 (d, J=1.8 Hz, 1H), 6.60 (dd, J=8.1 , 1.5 Hz, 1H), 4.08 (br. s., 2H), 3.42 (s, 3H), 2.58 (d, J=7.0 Hz, 4H), 2.52 – 2.42 (m, 1H), 2.09 – 1.98 (m, 1H), 1.79 – 1.59 (m, 3H), 1.32 – 1.22 (m, 1H), 0.94 – 0.84 (m, 12H)
Enantiomer 1 was prepared following the urea formation and saponification procedure in racemic example 1 method A. Chiral analytical analysis verified it was enantiomer 1 with 98.1% ee (Method J).
Example 1 – Method C Enantiomer 2
(lS,2R)-2-(4-(diisobutylamino)-3-(3-(p-tolyl)ureido)phenyl)cyclopropanecarboxylic acid

Example 1 Enantiomer 2 was prepared following the procedure for Example 1 enantiomer 1 method C using diastereomer 2 instead of diastereomer 1. Chiral analytical analysis verified it was enantiomer 2 with 94.0% ee (Method J).
PAPER
https://pubs.acs.org/doi/10.1021/acs.oprd.8b00171
Development of a Scalable Synthesis of BMS-978587 Featuring a Stereospecific Suzuki Coupling of a Cyclopropane Carboxylic Acid
 , Eric M. Simmons‡ , and Rajappa Vaidyanathan*†
, Eric M. Simmons‡ , and Rajappa Vaidyanathan*† 
A modified synthetic route to BMS-978587 was developed featuring a chemoselective nitro reduction and a stereospecific Suzuki coupling as the key bond formation steps. A systematic evaluation of the reaction conditions led to the identification of a robust catalyst/ligand/base combination to reproducibly effect the Suzuki reaction on large scale. The modified route avoided several challenges with the original synthesis and furnished the API in high overall yield and purity without recourse to chromatography.
(1R,2S)-2-[4-(Di-isobutylamino)-3-(3-(p-tolyl)ureido)phenyl] Cyclopropanecarboxylic Acid (1)
REF
(a) Balog, J. A.; Huang, A.; Chen, B.; Chen, L.; Seitz, P.; Hart, A. C.; Markwalder, J. A. Preparation of cycloalkylaryl amide compounds as indoleamine 2,3-dioxygenase and therapeutic uses thereof, PCT Int. Appl. 2014, WO 2014150677A1 20140925.
(b) Balog, J. A.; Cherney, E. C.; Guo, W.; Huang, A.; Markwalder, J. A.; Seitz, S. P.; Shan, W.; Williams, D. K.; Murugesan, N.; Nara, S.Jethanand; Preparation of benzenediamine derivatives as inhibitors of indoleamine 2,3-dioxygenase for the treatment of cancer, PCT Int. Appl. 2016, WO 2016161269A1 20161006.
////////////BMS-978587, IDO-IN-4, 1629125-65-0, CS-5086, BMS978587, BMS 978587
OC(=O)[C@@H]3C[C@@H]3c2cc(NC(=O)Nc1ccc(C)cc1)c(cc2)N(CC(C)C)CC(C)C