














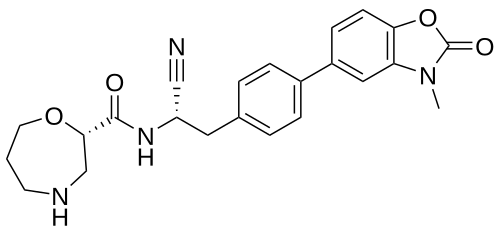
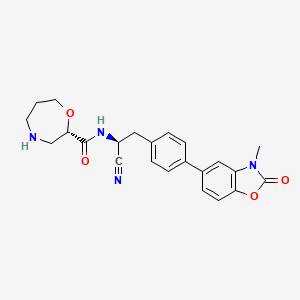

Brensocatib
WeightAverage: 420.469
Monoisotopic: 420.179755269
Chemical FormulaC23H24N4O4
- AZD7986
- CAS 1802148-05-5
- INS1007
- AZD 7986
- WHO 11097
(2S)-N-[(1S)-1-cyano-2-[4-(3-methyl-2-oxo-1,3-benzoxazol-5-yl)phenyl]ethyl]-1,4-oxazepane-2-carboxamide
- 1,4-Oxazepine-2-carboxamide, N-((1S)-1-cyano-2-(4-(2,3-dihydro-3-methyl-2-oxo-5-benzoxazolyl)phenyl)ethyl)hexahydro-, (2S)-
- (2S)-N-((1S)-1-cyano-2-(4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide
- (2S)-N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide
- Brensocatib [USAN]
- (S)-N-((S)-1-cyano-2-(4-(3-methyl-2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide
- UNII-25CG88L0BB
- (2S)-N-[(1S)-1-cyano-2-[4-(3-methyl-2-oxo-1,3-benzoxazol-5-yl)phenyl]ethyl]-1,4-oxazepane-2-carboxamide
FDA 8/12/2025. Brinsupri, To treat non-cystic fibrosis bronchiectasis
Brensocatib is an investigational new drug that is being evaluated to treat bronchiectasis.[1] It is a dipeptidyl-peptidase I (also known as cathepsin C) inhibitor.[2]
A phase 3 clinical trial, known as the ASPEN trial, was conducted to evaluate the safety and efficacy of brensocatib in patients with non-cystic fibrosis bronchiectasis.[3] Brensocatib tablets (Brinsupri) by Insmed Inc. was approved by the FDA in August 2025 after it received breakthrough therapy designation and was reviewed on a priority timeline.
Brensocatib is an orally bioavailable, small molecule, reversible inhibitor of dipeptidyl peptidase 1 (DPP1), with potential anti-inflammatory activity. Upon oral administration, brensocatib reversibly binds to and inhibits the activity of DPP1, thereby inhibiting the activation of neutrophil serine proteases (NSPs), including neutrophil elastase (NE), during neutrophil maturation. This inhibits the activity of NSPs, and may prevent lung inflammation and injury and improve lung function associated with NSPs-induced respiratory diseases. NSPs, serine proteases released by neutrophils during inflammation, is upregulated in a number of respiratory diseases.
SYN
J. Med. Chem. 2016, 59, 9457–9472, DOI: 10.1021/acs.jmedchem.6b01127
https://www.thieme-connect.com/products/ejournals/pdf/10.1055/s-0040-1719365.pdf
SYN
Brensocatib is now a clinical candidate to impair proteasedriven tissue degradation in COVID-19 (B. Korkmaz,
A. Lesner, S. Marchand-Adam, C. Moss, D. E. Jenne
J. Med. Chem. 2020, 63, 13258).
PAT
https://patents.google.com/patent/US9522894B2/en
A compound according to claim 1
which is (2S)—N-{(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide

EXAMPLESExample 1(2S)—N-[(1S)-1-Cyano-2-(4′-cyanobiphenyl-4-yl)ethyl]-1,4-oxazepane-2-carboxamide

i) tert-Butyl(2S)-2-{[(1S)-1-cyano-2-(4′-cyanobiphenyl-4-yl)ethyl]carbamoyl}-1,4-oxazepane-4-carboxylate
2-Pyridinol-1-oxide (0.155 g, 1.4 mmol), TEA (0.36 g, 3.6 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (0.268 g, 1.4 mmol) were added to a solution of (2S)-4-(tert-butoxycarbonyl)-1,4-oxazepane-2-carboxylic acid (Intermediate 3, 0.294 g, 1.2 mmol) in DCM (15 mL). After 20 min
4′-[(2S)-2-amino-2-cyanoethyl]biphenyl-4-carbonitrile (Intermediate 1, 0.296 g, 1.2 mmol) was added and the mixture was stirred for 3 h and allowed to stand at rt for 18 h. The mixture was heated at 40° C. for 4 h before water (15 mL) was added. After 10 min the DCM was dried (phase separating cartridge) and evaporated under reduced pressure. The resultant yellow oil was purified by silica gel column chromatography to give the subtitled compound (0.29 g, 52%). Used without further purification in the next step.ii) (2S)—N-[(1S)-1-Cyano-2-(4′-cyanobiphenyl-4-yl)ethyl]-1,4-oxazepane-2-carboxamide
Prepared according to procedure in Method A step ii) using tert-butyl(2S)-2-{[(1S)-1-cyano-2-(4′-cyanobiphenyl-4-yl)ethyl]carbamoyl}-1,4-oxazepane-4-carboxylate to afford the title compound as a white solid (60 mg, 28%).
1H NMR (400 MHz, CDCl3): δ 7.77-7.65 (m, 4H), 7.62-7.57 (m, 2H), 7.40 (d, 2H), 7.11 (d, 1H), 5.18-5.11 (m, 1H), 4.19-4.14 (m, 1H), 4.06-3.96 (m, 2H), 3.75-3.69 (m, 1H), 3.56-3.48 (m, 2H), 3.18-3.05 (m, 3H), 2.95-2.90 (m, 1H), 2.70 (ddd, 1H) (1 exchangeable proton not observed).
LCMS (10 cm_ESCI_Formic_MeCN) tR 2.57 (min) m/z 375 (MH+).Example 2(2S)—N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide

i) tert-Butyl(2S)-2-({(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}carbamoyl)-1,4-oxazepane-4-carboxylate

N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (468 mg, 2.44 mmol) and 2-pyridinol 1-oxide (271 mg, 2.44 mmol) were added to a solution of (2S)-4-(tert-butoxycarbonyl)-1,4-oxazepane-2-carboxylic acid (Intermediate 3, 490 mg, 2.0 mmol) in DCM (15 mL). The reaction was stirred at rt for 30 min before the addition of (2S)-2-amino-3-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]propanenitrile (Intermediate 2, 586 mg, 2.0 mmol) and DiPEA (1.79 mL, 10 mmol). The reaction was stirred at rt for 18 h before transferring to a separating funnel. The mixture was washed with 2 M hydrochloric acid, saturated sodium hydrogen carbonate solution and brine. The organic extract was run through a hydrophobic frit/phase separator and concentrated under reduced pressure. The crude material was purified by silica gel column chromatography eluting with 0-60% EtOAc in iso-hexane to afford the subtitled compound as an oil (457 mg, 44%). 1H NMR (400 MHz, CDCl3): δ 7.63-7.52 (m, 2H), 7.38 (d, 2H), 7.36-7.24 (m, 2H), 7.35-6.98 (m, 2H), 5.18 (t, 1H), 4.22-3.97 (m, 2H), 3.76-3.67 (m, 0.5H), 4.10-2.94 (m, 4.5H), 3.35-3.26 (m, 1H), 3.24-3.04 (m, 3H), 2.06-1.82 (m, 2H), 1.47 (s, 10H).ii) (2S)—N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide
tert-Butyl(2S)-2-({(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}carbamoyl)-1,4-oxazepane-4-carboxylate (457 mg, 0.85 mmol) was dissolved in formic acid (3 mL) and heated at 50° C. for 10 min on a pre-heated stirrer hotplate. After this time the reaction was concentrated under reduced pressure, dissolved in DCM and washed with saturated sodium hydrogen carbonate solution. The organic extract was run through a hydrophobic frit/phase separator and concentrated under reduced pressure. The resultant foam was purified by silica gel column chromatography eluting with 0-5% methanolic ammonia (7 N) in DCM to afford the title compound as solid material (230 mg, 64%).
1H NMR (400 MHz, CDCl3): δ 7.59-7.51 (m, 2H), 7.39 (dd, 2H), 7.33-7.23 (m, 3H), 7.14 (d, 1H), 5.23-5.12 (m, 1H), 4.12-4.06 (m, 1H), 4.05-3.95 (m, 1H), 3.81-3.71 (m, 1H), 3.46 (s, 3H), 3.34-3.26 (m, 1H), 3.19-3.00 (m, 3H), 2.99-2.82 (m, 2H), 1.92-1.77 (m, 2H) (one exchangeable proton not observed).
LCMS (10 cm_ESCI_Formic_MeCN) tR 2.48 (min) m/z 375 (MH+).Example 2Alternative Synthesis(2S)—N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamidei) 5-Chloro-1,3-benzoxazol-2(3H)-one

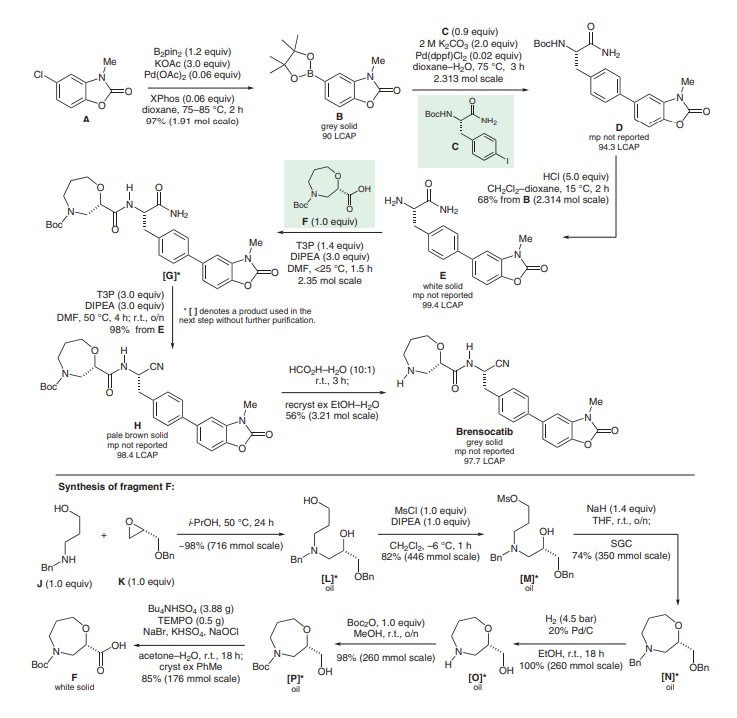
To a solution of 2-amino-4-chlorophenol (400 g, 2.79 mol) in 2-MeTHF (6 L) was added CDI (497 g, 3.07 mol) under N2 (exotherm 11.0° C.-22.0° C.). The reaction mixture was heated at reflux for 1 h. The mixture was cooled to rt, washed with 2 M HCl(aq) (6 L), 8% NaHCO3(aq) (6 L) and brine (3 L). The organic layer was dried over MgSO4, filtered and evaporated. This gave the product as a pale brown solid (456.1 g, 97% yield, LC purity >99%).
1H NMR (270 MHz, DMSO-d6): δ 12.0-11.5 (br s, 1H), 7.31 (d, 1H), 7.12 (m, 2H).
LCMS (5 cm_ESCI, aq. formic acid_methanol) tR 3.87 (min) m/z 169.8 (MH+).ii) 5-Chloro-3-methyl-1,3-benzoxazol-2(3H)-one

To a solution of 5-chloro-1,3-benzoxazol-2(3H)-one (stage i) (1111.8 g, 6.56 mol) in DMF (4.12 L) was added Cs2CO3 (2136.4 g, 6.56 mol) maintaining the temperature between 0-5° C. MeI (450 ml, 7.21 mol) was then added slowly maintaining the temperature between 0-5° C. The reaction mixture was allowed to warm-up to rt and stirred overnight. The mixture was cooled to 0-5° C. and H2O (4.12 L) was added slowly. The reaction mixture was then warmed to rt and stirred for 15 min. The solids were filtered off and washed with water (4×980 ml). The filter cake was dried under vacuum at 55° C. overnight (1149.9 g, 96% yield, LC purity >99%, H2O: (Karl Fischer) 0.1%).
1H NMR (270 MHz, DMSO-d6): δ 7.45 (d, 1H), 7.35 (d, 1H), 7.15 (dd, 1H), 3.35 (s, 3H). LCMS (5 cm_ESCI_aq. formic acid_methanol) tR 4.13 (min) m/z 183.8 (M+).iii) 3-Methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-benzoxazol-2(3H)-one

A solution of 5-chloro-3-methyl-1,3-benzoxazol-2(3H)-one (stage ii)) (350 g, 1.91 mol), B2pin2 (581.0 g, 2.29 mol) and KOAc (561.3 g, 5.72 mol) was vacuum degassed and purged with N2 (×3). Pd(OAc)2 (12.9 g, 57.2 mmol) and XPhos (54.6 g, 114 mmol) were added and the mixture was vacuum degassed and purged with N2 (×3). The mixture was heated to 75° C. A large exotherm was observed at ˜70° C. which warmed-up the mixture to reflux (100° C.). The reaction mixture was stirred for 1 h with no heating. HPLC analysis indicated 2.5% of the starting material remaining therefore the mixture was heated at 85° C. for 1 h. At this stage, no further change was observed. Additional portions of B2pin2 (14.6 g, 57.2 mmol), KOAc (5.7 g, 57.2 mmol), Pd(OAc)2 (12.9 g, 57.2 mmol) and XPhos (27.3 g, 57.2 mmol) were added and the mixture was stirred for 1 h at 75° C. HPLC analysis showed no starting material remaining. The mixture was cooled to rt, filtered through a pad of Celite (501 g) and the cake was washed with EtOAc (2240 ml). The filtrate was combined with two other batches prepared in the same way (2×350 g) and evaporated. This gave 1865.1 g of the product as a grey solid (97% yield, 90.0% pure by LC, 82±2% pure by 1H NMR (DMSO-d6) assay vs TCNB).
1H NMR (270 MHz, DMSO-d6): δ 7.40-7.50 (m, 2H), 7.30 (d, 1H), 3.40 (s, 3H), 1.30 (s, 12H).
LCMS (5 cm_ESCI_aq. formic acid_methanol_) tR 4.91 (min) m/z 276.1 (MH+).iv) Nα-(tert-Butoxycarbonyl)-4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)-L-phenylalaninamide

To a suspension of 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-benzoxazol-2(3H)-one (stage iii)) (859 g, 700 g active, 2.544 mol) and tert-butyl (S)-1-carbamoyl-2-(4-iodophenypethylcarbamate (prepared according to the procedure in WO 2009/074829 p. 47), (903 g, 2.313 mol) in dioxane (4.1 L) was added 2 M K2CO3 (2.3 L). The suspension was vacuum degassed and purged with N2 (×3). Pd(dppf)Cl2.DCM (28.33 g, 0.0347 mol) was added and the reaction mixture was heated at 75° C. for 3 h. The mixture was cooled to rt and diluted with water (6.4 L). The suspension was stirred at rt overnight; the solid was filtered off and washed with water (3×1 L). The product was dried at 45° C. for 3 days (1269.1 g, yield 133%—by 1H NMR contains pinacol related impurity and dioxane, LC 94.3% pure, H2O: (Karl Fischer) 3.35%).
1H NMR (270 MHz, DMSO-d6): δ 7.62-7.34 (m, 7H), 7.04 (brs, 2H), 6.86 (d, 1H) 4.12 (m, 1H), 3.40 (s, 3H), 3.00 (dd, 1H), 2.78 (dd, 1H), 1.30 (s, 9H).
LCMS (5 cm_ESI_Water_MeCN) tR 4.51 (min) m/z 312 (MH+).v) 4-(3-Methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)-L-phenylalaninamide

To a very thick suspension of Nα-(tert-butoxycarbonyl)-4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)-L-phenylalaninamide (stage iv)) (1269 g, active 952 g assumed 100% conversion at stage iv), 2.3138 mol) in DCM (2.1 L) under N2 was added dropwise 4.1 M HCl in dioxane (2.7 L, 11.06 mol) over 1 h maintaining the temperature at 15° C. (suspension became more mobile after addition of approx. 0.5 L of 4.1 M HCl dioxane). After 2 h, the mixture was diluted with water (5.6 L) and stirred for 30 min at rt. The mixture was then filtered through a pad of Celite (500 g) to remove undissolved material—very slow filtration; the Celite was checked for product by LC. The pad was washed with water (400 ml). The layers DCM/dioxane-water were separated. The aqueous layer was cooled to ˜5° C. and 35% NH3 (aq) (700 ml) was added slowly to achieve pH=9-10. The suspension was stirred overnight then the product was filtered off and washed with water (3×400 ml). The product was dried at 45° C. in vacuo for 2 days (off white solid, 489.4 g, 68% yield over two stages, 99.4% pure by LC, >99% EP, 98±2% pure by 1H NMR assay vs TCNB in DMSO, H2O: (Karl Fischer) 0.92%).
1H NMR (270 MHz, DMSO-d6): δ 7.59-7.30 (m, 7H), 6.98 (brs, 1H), 3.36 (m, 4H), 2.95 (dd, 1H), 2.67 (dd, 1H) 1.86 (brs, 2H).
LCMS (5 cm_ESI_Water_MeCN) tR 2.76 (min) m/z 312 (MH+).vi) tert-Butyl(2S)-2-({(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}carbamoyl)-1,4-oxazepane-4-carboxylate

To a solution of 4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)-L-phenylalaninamide (stage v)) (756 g, active 733 g, 2.354 mol) and (2S)-4-(tert-butoxycarbonyl)-1,4-oxazepane-2-carboxylic acid (577 g, 2.354) (Intermediate 3) in DMF (3 L) was added DiPEA (1230 ml, 7.062 mol) under N2. T3P in DMF (50% w/w, 1924 ml, 3.296 mol) was added dropwise over 1.5 h maintaining the temperature<25° C. After 30 min, LC completion check indicated completion of the coupling reaction. DiPEA (1230 ml, 7.062 mol) was then added and the reaction mixture was heated to 50° C. T3P in DMF (50% w/w, 3986 ml, 6.827 mol) was added portionwise over 1 h (no exotherm observed). The reaction mixture was stirred at 50° C. for 4 h and then at rt overnight. The mixture was cooled to 10° C., diluted with 2-MeTHF (4 L) and water (5.6 L, exothermic). The layers were separated and the aqueous layer was extracted with 2-MeTHF (2×4 L). The combined organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure. This delivered the product as a pale brown solid in 98% yield (1242 g (active 1205 g), corrected yield 98%, LC purity 98.4%, 1H NMR assay vs TCNB 97±2%, main impurities by 1H NMR: 2-MeTHF 1.9%, DMF 0.6%).vii) (2S)—N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide
A solution of tert-butyl(2S)-2-({(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}carbamoyl)-1,4-oxazepane-4-carboxylate (stage vi)) (1776 g, active 1671 g, 3.210 mol) in formic acid/water (4.2 L/440 ml) was stirred on a buchi at 35-37° C. under reduced pressure (300-500 mbar). After 3 h, LCMS completion check indicated 93.95% of the product and 0.5% of the starting material. The mixture was concentrated (4 h) to give an oily residue. The residue was dissolved in water (4.4 L) and washed with TBME (2.2 L). The aqueous layer was vigorously stirred and treated with NH3(aq) (1.8 L) at <25° C. to achieve pH=9-10. The mixture was stirred at rt for 3 h. The solid was filtered off and washed with water (3×1 L). The filter cake was dried at 45° C. overnight. This gave the product as a pale brown solid (1498 g, active 1333 g, LC 91.5%, 1H NMR assay vs TCNB 89±2%, H2O: (Karl Fischer) 4.63%).
The crude product was re-crystallised from EtOH/H2O in two batches (2×747 g).
Batch A: The crude product (747 g) was dissolved in EtOH (8 L) at reflux under N2. Water (1.6 L) was added slowly. The mixture was hot filtered (65° C.) to remove black particles (filtrate temperature
50° C.) and then stirred at 40° C. overnight. The suspension was cooled to 10° C. over 4 h and held at that temperature for 3 h. The product was filtered off and washed with EtOH/H2O (8:2, 3×500 ml) then water (3×500 ml). The filter cake was dried at 45° C. overnight (473 g, 97.7% pure by LC, Pd level 71.4 ppm).
Batch B gave 436 g of the product (95.8% pure by LC, Pd level 65.8 ppm).
The liquors from both batches were combined and concentrated to ˜8 L. The liquors were left overnight at rt. The solids were filtered off and washed with EtOH/H2O (8:2, 3×400 ml) then water (3×400 ml). The product was dried at 45° C. overnight. This gave additional 88 g of the product (LC purity 95.0%).
The products (LC purity of the blend 95.69%) were re-crystallised from EtOH/H2O in two batches (Batch C: 520 g, Batch D: 520 g).
Batch C: The crude product (520 g) was dissolved in EtOH (6.24 L) at reflux under N2. Water (1248 ml) was added slowly. The mixture was allowed to cool down to 40° C. (3 h), seeded with 0.5 g of the title compound and stirred at 40° C. for 10 h. The mixture was then cooled to 26° C. over 7 h. The resulting suspension was cooled to 10° C. and stirred at that temperature for 6 h. The product was filtered off, washed with EtOH/water (8:2, 3×500 ml) and water (3×500 ml). The filter cake was dried at 45° C. for 2 d. The product was obtained as a grey solid (418 g, yield ˜56%, LCMS purity 97.5%, chiral LC
100%, 1H NMR (DMSO-d6) assay vs TCNB
100±2%).
Batch D: 418 g, yield 56%, LCMS purity 97.5%, chiral LC
100%, 1H NMR (DMSO-d6) assay vs TCNB
100±2%
The product was blended with the material from an intermediate scale reaction performed in the same way and re-analysed (968 g, LC purity 98.04%, chiral LC
100%, 1H NMR assay vs TCNB 99±2%, 0.35% EtOH by 1H NMR, H2O: (Karl Fischer) 4.58%, Pd 57.6 ppm, XRPD (X-ray powder diffraction) Form A.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- “Brensocatib – Insmed”. AdisInsight. Springer Nature Switzerland AG.
- Chalmers JD, Usansky H, Rubino CM, Teper A, Fernandez C, Zou J, et al. (October 2022). “Pharmacokinetic/Pharmacodynamic Evaluation of the Dipeptidyl Peptidase 1 Inhibitor Brensocatib for Non-cystic Fibrosis Bronchiectasis”. Clinical Pharmacokinetics. 61 (10): 1457–1469. doi:10.1007/s40262-022-01147-w. PMC 9553789. PMID 35976570.
- Chalmers JD, Burgel PR, Daley CL, De Soyza A, Haworth CS, Mauger D, et al. (April 2025). “Phase 3 Trial of the DPP-1 Inhibitor Brensocatib in Bronchiectasis”. The New England Journal of Medicine. 392 (16): 1569–1581. doi:10.1056/NEJMoa2411664. PMID 40267423.
| Clinical data | |
|---|---|
| Other names | AZD7986; INS1007 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1802148-05-5 |
| PubChem CID | 118253852 |
| IUPHAR/BPS | 9412 |
| DrugBank | DB15638 |
| ChemSpider | 67896269 |
| UNII | 25CG88L0BB |
| KEGG | D12120 |
| ChEMBL | ChEMBL3900409 |
| Chemical and physical data | |
| Formula | C23H24N4O4 |
| Molar mass | 420.469 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
////////Brensocatib, APPROVALS 2025, FDA 2025, Brinsupri, non-cystic fibrosis, AZD7986, 1802148-05-5, INS1007, AZD 7986, WHO 11097