














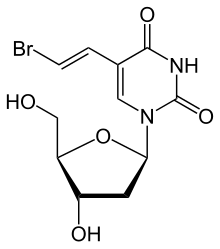
Brivudine
ブリブジン;
| Formula |
C11H13BrN2O5
|
|---|---|
| CAS |
69304-47-8
|
| Mol weight |
333.1353
|
Brivudine (trade names Zostex, Mevir, Brivir, among others) is an antiviral drug used in the treatment of herpes zoster (“shingles”). Like other antivirals, it acts by inhibiting replication of the target virus.
Medical uses
Brivudine is used for the treatment of herpes zoster in adult patients. It is taken orally once daily, in contrast to aciclovir, valaciclovir and other antivirals.[1] A study has found that it is more effective than aciclovir, but this has been disputed because of a possible conflict of interest on part of the study authors.[2]
Contraindications
The drug is contraindicated in patients undergoing immunosuppression (for example because of an organ transplant) or cancer therapy, especially with fluorouracil (5-FU) and chemically related (pro)drugs such as capecitabine and tegafur, as well as the antimycotic drug flucytosine, which is also related to 5-FU. It has not been proven to be safe in children and pregnant or breastfeeding women.[1]
Adverse effects
The drug is generally well tolerated. The only common side effect is nausea (in 2% of patients). Less common side effects (<1%) include headache, increased or lowered blood cell counts (granulocytopenia, anaemia, lymphocytosis, monocytosis), increased liver enzymes, and allergic reactions.[1]
Interactions
Brivudine interacts strongly and in rare cases lethally with the anticancer drug fluorouracil (5-FU), its prodrugs and related substances. Even topically applied 5-FU can be dangerous in combination with brivudine. This is caused by the main metabolite, bromovinyluracil (BVU), irreversibly inhibiting the enzyme dihydropyrimidine dehydrogenase (DPD) which is necessary for inactivating 5-FU. After a standard brivudine therapy, DPD function can be compromised for up to 18 days. This interaction is shared with the closely related drug sorivudine which also has BVU as its main metabolite.[1][3]
There are no other relevant interactions. Brivudine does not significantly influence the cytochrome P450 enzymes in the liver.[1]
Pharmacology
Spectrum of activity
The drug inhibits replication of varicella zoster virus (VZV) – which causes herpes zoster – and herpes simplex virus type 1 (HSV-1), but not HSV-2 which typically causes genital herpes. In vitro, inhibitory concentrations against VZV are 200- to 1000-fold lower than those of aciclovir and penciclovir, theoretically indicating a much higher potency of brivudine. Clinically relevant VZV strains are particularly sensitive.[4]
Mechanism of action
Brivudine is an analogue of the nucleoside thymidine. The active compound is brivudine 5′-triphosphate, which is formed in subsequent phosphorylations by viral (but not human) thymidine kinase and presumably by nucleoside-diphosphate kinase. Brivudine 5′-triphosphate works because it is incorporated into the viral DNA, but then blocks the action of DNA polymerases, thus inhibiting viral replication.[1][4]
Pharmacokinetics
Brivudine is well and rapidly absorbed from the gut and undergoes first-pass metabolism in the liver, where the enzyme thymidine phosphorylase[5] quickly splits off the sugar component, leading to a bioavailability of 30%. The resulting metabolite is bromovinyluracil (BVU), which does not have antiviral activity. BVU is also the only metabolite that can be detected in the blood plasma.[1][6]
Highest blood plasma concentrations are reached after one hour. Brivudine is almost completely (>95%) bound to plasma proteins. Terminal half-life is 16 hours; 65% of the substance are found in the urine and 20% in the faeces, mainly in form of an acetic acid derivative (which is not detectable in the plasma), but also other water-soluble metabolites, which are urea derivatives. Less than 1% is excreted in form of the original compound.[1]
-
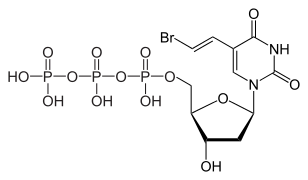
Brivudine 5′-triphosphate, the active metabolite
-

Bromovinyluracil (BVU), the main inactive metabolite
-

The acetic acid derivative predominantly found in urine



Chemistry
The molecule has three chiral carbon atoms in the deoxyribose (sugar) part all of which have defined orientation; i.e. the drug is stereochemically pure.[1] The substance is a white powder.
Manufacturing
Main supplier is Berlin-Chemie, now part of Italy’s Menarini Group. In Central America is provided by Menarini Centro America and Wyeth.
History
The substance was first synthesized by scientists at the University of Birmingham in the UK in 1976. It was shown to be a potent inhibitor of HSV-1 and VZV by Erik De Clercq at the Rega Institute for Medical Research in Belgium in 1979. In the 1980s the drug became commercially available in East Germany, where it was marketed as Helpin by a pharmaceutical company called Berlin-Chemie. Only after the indication was changed to the treatment of herpes zoster in 2001 did it become more widely available in Europe.[7][8]
Brivudine is approved for use in a number of European countries including Austria, Belgium, Germany, Greece, Italy, Portugal, Spain and Switzerland.[9]
Etymology
The name brivudine derives from the chemical nomenclature bromo–vinyl–deoxyuridine or BVDU for short. It is sold under trade names such as Bridic, Brival, Brivex, Brivir, Brivirac, Brivox, Brivuzost, Zerpex, Zonavir, Zostex, and Zovudex.[9]
Research
A Cochrane Systematic Review examined the effectiveness of multiple antiviral drugs in the treatment of herpes simplex virus epithelial keratitis. Brivudine was found to be significantly more effective than idoxuridine in increasing the number of successfully healed eyes of participants.[10]
PATENT
EP-03792271
https://patentscope.wipo.int/search/en/detail.jsf?docId=EP320237831&_cid=P11-KMRHYQ-39914-1
Process for preparing brivudine, useful for treating herpes zoster infection and cancer (eg pancreatic cancer). Also claims novel intermediate of brivudine. Brivudine is an antiviral drug approved under the brand name Zostex®. Represents the first patenting to be seen from Aurobindo that focuses on brivudine.
Brivudine is chemically known as 5-[(lE)-2-bromoethenyl]-2′-deoxyuridine. Brivudine is an analogue of the nucleoside thymidine with high and selective antiviral activity against varicella zoster virus and herpes simplex virus. Brivudine is an antiviral drug approved under the brand name Zostex® for treatment of herpes zoster. Brivudine is also useful to inhibit the upregulation of chemoresistance genes (Mdr1 and DHFR) during chemotherapy. Overall, the gene expression changes associated with Brivudine treatment result in the decrease or prevention of chemoresistance. In addition, it has been shown to enhance the cytolytic activity of NK-92 natural killer cells towards a pancreatic cancer cell line in vitro.
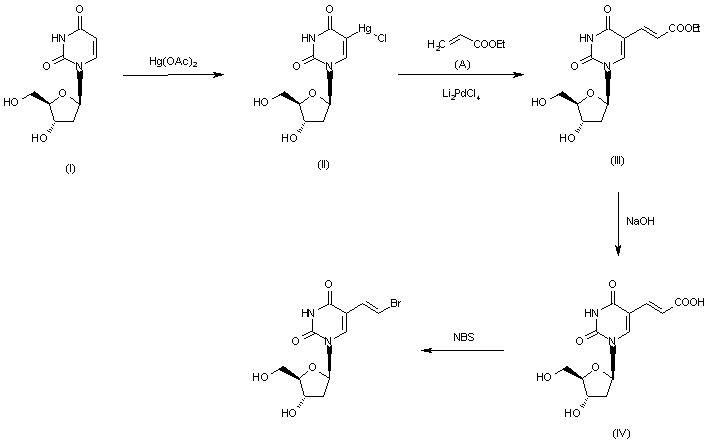
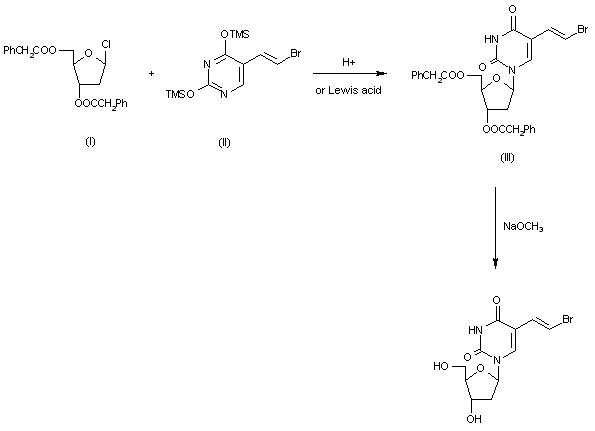
[0003] Brivudine (I) is disclosed first time in DE 2915254. This patent discloses a process for the preparation of Brivudine by coupling E-5-(2-bromovinyl) uracil with 1-chloro-2-deoxy-3,5-di-O-p-toluoyl-α-D-erythro-pentofuranose to obtain E-5-(2-bromovinyl)-3′,5′-di-O-p-toluoyl-2′-deoxyuridine as a mixture of α and β isomers. This mixture was subjected to chromatographic purification to obtain pure β-isomer. In the subsequent stage E-5-(2-bromovinyl)-3′,5′-di-O-p-toluoyl-2′-deoxyuridine was treated with sodium methoxide to yield Brivudine. The process is depicted in the below as Scheme I:
[0004] The major disadvantages associated with the process disclosed in DE 2915254 includes the use of expensive starting material, formation of unwanted excess of α-isomer. The undesired α-isomer result in a final product of low purity, making chromatographic purification methods not feasible at an industrial scale. Additionally, the process involves the use of bromine for the synthesis of E-5-(2-bromovinyl)uracil, which is a well known carcinogen.
[0005] GB 2125399 describe another process for the preparation of Brivudine involves the bromination and simultaneous dehydrohalogenation of 5-ethyl-2′-deoxyuridine in the presence of halogenated hydrocarbon solvent. The process is depicted in the below as Scheme – II:
[0006] The major disadvantages associated with the process disclosed in GB 2125399 includes the use of bromine for bromination, which make the process carcinogenic and the use of halogenated solvents like chloroform, carbon tetrachloride and dichloroethane for bromination makes the process environmentally hazardous.
[0007] US 2010298530 A1 discloses a process for the preparation of Brivudine by coupling 5-Iodo deoxyuridine with methyl acrylate in presence of palladium acetate to form (E)-5-(carbomethoxyvinyl)-2′-deoxyuridine, which is hydrolyzed with sodium hydroxide solution to obtain (E)-5-(carboxyvinyl)-2′-deoxyuridine, which undergoes bromination by using N-Bromo succinimide [NBS] to obtain Brivudine. The process is depicted in the below as Scheme – III:
[0008] The major disadvantages associated with the process disclosed in US 2010298530 A1 includes the use of expensive palladium acetate as catalyst and chromatographic purification method not feasible at an industrial scale. In addition, the above process involves the use of methyl acrylate and the process liberates iodine, which are highly carcinogenic. It makes the process environment unfriendly.
[0009] The inventors of the present invention found an alternative route to prepare Brivudine (I), which is industrial feasible, can avoid the use of potentially hazardous, expensive chemicals and to minimize the formation of undesired α-isomer and the other process related impurities. The present invention directed towards a process for the preparation of Brivudine of Formula – I with high purity and high yield.
EXAMPLE-1:
PREPARATION OF URIDINE ACRYLIC ACID
[0026] Trimethylchlorosilane (0.6 ml, 5 mmol) was added to the suspension of uracil acrylic acid (6.35g, 34.9 mmol) in hexamethyldisilazane (70 ml) and resulting mixture was refluxed till the clear solution was obtained. Hexamethyldisilazane was evaporated under vacuum and further co-evaporated with o-xylene to remove the traces of hexamethyldisilazane to yield viscous oily silylated uracil acrylic acid. The silylated uracil acrylic acid was dissolved in dichloromethane (100 ml) under nitrogen atmosphere, cooled at 0-10 °C. Anhydrous zinc chloride (0.63 grams, 4.6 mmol) and chloro-sugar (10 grams, 23.2 mmol) were added to the above solution. The reaction was monitored by qualitative HPLC and was essentially completed in 3 hours. After completion of the reaction dichloromethane was evaporated under vacuum. Methyl tert-butyl ether (100 ml) was added and stirred for 1 hour at 40-45 °C. The product was isolated after filtration at 25-30 °C. HPLC analysis showed the complete consumption of chloro-sugar and the ratio β/α = 98.
Yield: 75%
EXAMPLE-2:
PREPARATION OF DIBENZOYL BRIVUDINE
[0027] Uridine acrylic acid (5.0 grams, 8.69 mmol) was suspended in a mixture of tetrahydrofuran (45.0 ml) and water (5.0 ml). Potassium acetate (0.93 grams, 9.56 mmol) and N-bromosuccinamide (1.70 grams, 9.56 mmol) was added to the suspension and the resulting mixture was stirred for 2 hours at 25-30 °C. The solvent was removed under reduced pressure to the dryness and methanol (50 ml) was poured, suspension was stirred for 1 hour at 25-30 °C. The product was isolated after filtration.
Yield: 55%
EXAMPLE-3:
PREPARATION OF BRIVUDINE (FORMULA – I)
[0028] Dibenzoyl Brivudine (6 grams, 9.83 mmol) was suspended in methanol (30 ml) at 20-30 °C. A solution of 25 % w/w sodium methoxide (2.76 grams, 12.78 mmol) in methanol was added to the suspension and was allowed for 1hour at the same temp. The reaction was monitored by qualitative HPLC. Methanol was evaporated under vacuum and resulting residue was dissolved in water (25 ml). The aqueous mass was washed with methylene dichloride (2×20 ml) and the product was isolated from water at pH 2-3.
Yield: 85%
SYN
http://sioc-journal.cn/Jwk_yjhx/EN/10.6023/cjoc201410034

References
- ^ Jump up to:a b c d e f g h i Jasek W, ed. (2007). Austria-Codex (in German) (62nd ed.). Vienna: Österreichischer Apothekerverlag. pp. 5246–8. ISBN 978-3-85200-181-4.
- ^ “Brivudin (Zostex) besser als Aciclovir (Zovirax a.a.)?”. Arznei-telegramm (in German). 5/2007.
- ^ “UAW – Aus Fehlern lernen – Potenziell tödlich verlaufende Wechselwirkung zwischen Brivudin (Zostex) und 5-Fluoropyrimidinen” (PDF). Deutsches Ärzteblatt (in German). 103 (27). 7 July 2006.
- ^ Jump up to:a b Steinhilber D, Schubert-Zsilavecz M, Roth HJ (2005). Medizinische Chemie (in German). Stuttgart: Deutscher Apotheker Verlag. pp. 581–2. ISBN 3-7692-3483-9.
- ^ Desgranges C, Razaka G, Rabaud M, Bricaud H, Balzarini J, De Clercq E (December 1983). “Phosphorolysis of (E)-5-(2-bromovinyl)-2′-deoxyuridine (BVDU) and other 5-substituted-2′-deoxyuridines by purified human thymidine phosphorylase and intact blood platelets”. Biochemical Pharmacology. 32 (23): 3583–90. doi:10.1016/0006-2952(83)90307-6. PMID 6651877.
- ^ Mutschler E, Schäfer-Korting M (2001). Arzneimittelwirkungen (in German) (8 ed.). Stuttgart: Wissenschaftliche Verlagsgesellschaft. p. 847. ISBN 3-8047-1763-2.
- ^ De Clercq E (December 2004). “Discovery and development of BVDU (brivudin) as a therapeutic for the treatment of herpes zoster”. Biochemical Pharmacology. 68 (12): 2301–15. doi:10.1016/j.bcp.2004.07.039. PMID 15548377.
- ^ Tringali C, ed. (2012). Bioactive Compounds from Natural Sources (2nd ed.). CRC Press. p. 170.
- ^ Jump up to:a b International Drug Names: Brivudine.
- ^ Wilhelmus KR (January 2015). “Antiviral treatment and other therapeutic interventions for herpes simplex virus epithelial keratitis”. The Cochrane Database of Systematic Reviews. 1: CD002898. doi:10.1002/14651858.CD002898.pub5. PMC 4443501. PMID 25879115.
 |
|
| Clinical data | |
|---|---|
| Trade names | Zostex, Mevir, Brivir, many others |
| Other names | BVDU |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 30% |
| Protein binding | >95% |
| Metabolism | Thymidine phosphorylase |
| Metabolites | Bromovinyluracil |
| Elimination half-life | 16 hours |
| Excretion | 65% renal (mainly metabolites), 20% faeces |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C11H13BrN2O5 |
| Molar mass | 333.138 g·mol−1 |
| 3D model (JSmol) | |
| Specific rotation | +9°±1° |
| Density | 1.86 g/cm3 |
| Melting point | 165 to 166 °C (329 to 331 °F) (decomposes) |
|
|
|