














Casimersen
カシメルセン;
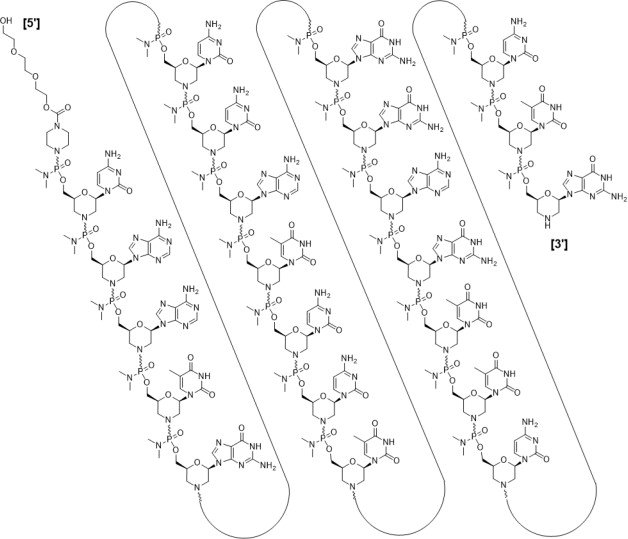
RNA, [P-deoxy-P-(dimethylamino)](2′,3′-dideoxy-2′,3′-imino-2′,3′-seco)(2’a→5′)(C-A-A-m5U-G-C-C-A-m5U-C-C-m5U-G-G-A-G-m5U-m5U-C-C-m5U-G), 5′-[P-[4-[[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]carbonyl]-1-piperazinyl]-N,N-dimethylphosphonamidate]
| Formula |
C268H424N124O95P22
|
|---|---|
| CAS |
1422958-19-7
|
| Mol weight |
7584.4307
|
FDA 2021/2/25 , Amondys 45, Antisense oligonucleotide
Treatment of Duchenne muscular dystrophy
Nucleic Acid Sequence
Sequence Length: 224 a 7 c 5 g 6 umodified
- Exon-45: NG-12-0064
- SRP-4045
- WHO 10354
Casimersen, sold under the brand name Amondys 45, is an antisense oligonucleotide medication used for the treatment of Duchenne muscular dystrophy (DMD) in people who have a confirmed mutation of the dystrophin gene that is amenable to exon 45 skipping.[1][2][3][4] It is an antisense oligonucleotide of phosphorodiamidate morpholino oligomer (PMO).[1]
The most common side effects include upper respiratory tract infections, cough, fever, headache, joint pain and throat pain.[2]
Casimersen was approved for medical use in the United States in February 2021,[1][2] and it is the first FDA-approved targeted treatment for people who have a confirmed mutation of the DMD gene that is amenable to skipping exon 45.[2]
Duchenne muscular dystrophy (DMD) is an X-linked recessive allelic disorder characterized by a lack of functional dystrophin protein, which leads to progressive impairment of ambulatory, pulmonary, and cardiac function and is invariably fatal. A related, albeit a less severe, form of muscular dystrophy known as Becker muscular dystrophy (BMD) is characterized by shortened and partially functional dystrophin protein production. Although corticosteroids effectively slow disease progression in both DMD and BMD patients, they do not address the underlying molecular pathogenesis.1,2,3
The application of antisense oligonucleotides in DMD patients with specific mutations allows for exon skipping to produce truncated BMD-like dystrophin proteins, which restore partial muscle function and slow disease progression.1,2,4,5,7 Casimersen is a phosphorodiamidate morpholino oligonucleotide (PMO); PMOs are oligonucleotides in which the five-membered ribofuranosyl ring is replaced with a six-membered morpholino ring, and the phosphodiester links between nucleotides are replaced with a phosphorodiamidate linkage.6,7 In this manner, PMOs are much less susceptible to endo- and exonucleases and exhibit drastically reduced metabolic degradation compared to traditional synthetic oligonucleotides.6 Casimersen is the most recent in a line of approved PMOs for treating DMD, including eteplirsen and viltolarsen. However, the specific mutations, and hence the precise exon skipping, targeted by each is different.
Casimersen was granted accelerated FDA approval on February 25, 2021, based on data showing an increase in dystrophin levels in skeletal muscle of patients treated with casimersen; this approval is contingent on further verification in confirmatory trials. Casimersen is currently marketed under the tradename AMONDYS 45™ by Sarepta Therapeutics, Inc.7
Casimersen is indicated for the treatment of Duchenne muscular dystrophy (DMD) in patients confirmed to have a DMD gene mutation amenable to exon 45 skipping. This indication represents an accelerated approval based on observed efficacy; continued approval for this indication may be contingent on the verification of safety and efficacy in a confirmatory trial.7
Medical uses
Casimersen is indicated for the treatment of Duchenne muscular dystrophy (DMD) in people who have a confirmed mutation of the DMD gene that is amenable to exon 45 skipping.[1][2]
History
Casimersen was evaluated in a double-blind, placebo-controlled study in which 43 participants were randomized 2:1 to receive either intravenous casimersen or placebo.[2] All participants were male, between 7 and 20 years of age, and had a genetically confirmed mutation of the DMD gene that is amenable to exon 45 skipping.[2]
The U.S. Food and Drug Administration (FDA) granted the application for casimersen fast track, priority review, and orphan drug designations.[2][5] The FDA granted the approval of Amondys 45 to Sarepta Therapeutics, Inc.[2]
Pharmacodynamics
Casimersen is an antisense phosphorodiamidate morpholino oligonucleotide designed to bind to exon 45 of the DMD pre-mRNA, preventing its inclusion in mature mRNA and allowing the production of an internally truncated dystrophin protein in patients who would normally produce no functional dystrophin. Due to the need for continuous alteration of mRNA splicing and its relatively short half-life, casimersen is administered weekly.7 Although casimersen is associated with mostly mild adverse effects, animal studies suggest a potential for nephrotoxicity, which has also been observed after administration of some oligonucleotides.4,7 Measurement of glomerular filtration rate before starting casimersen is advised. Serum cystatin C, urine dipstick, and urine protein-to-creatinine ratio should be measured before starting therapy. They should be measured monthly (urine dipstick) or every three months (serum cystatin C and urine protein-to-creatinine ratio) during treatment. Creatinine levels are not reliable in muscular dystrophy patients and should not be used. Any persistent alteration in kidney function should be further investigated.7
Mechanism of action
Duchenne muscular dystrophy (DMD) is an X-linked recessive allelic disorder that results in the absence of functional dystrophin, a large protein comprising an N-terminal actin-binding domain, C-terminal β-dystroglycan-binding domain, and 24 internal spectrin-like repeats.1,2,3 Dystrophin is vital for normal muscle function; the absence of dystrophin leads to muscle membrane damage, extracellular leakage of creatinine kinase, calcium influx, and gradual replacement of normal muscle tissue with fibrous and adipose tissue over time.1,2 DMD shows a characteristic disease progression with early functional complaints related to abnormal gait, locomotion, and falls that remain relatively stable until around seven years of age. The disease then progresses rapidly to loss of independent ambulatory function, ventilatory insufficiency, and cardiomyopathy, with death typically occurring in the second or third decade of life.1,2,3
The human DMD gene contains 79 exons spread over approximately 2.4 million nucleotides on the X chromosome.1 DMD is associated with a variety of underlying mutations, including exon duplications or deletions, as well as point mutations leading to nonsense translation through direct production of an in-frame stop codon, frameshift production of an in-frame stop codon, or aberrant inclusion of an intronic pseudo-exon with the concomitant production of an in-frame stop codon.1,2 In all cases, no functional dystrophin protein is produced. Becker muscular dystrophy (BMD) is a related condition with in-frame mutations that result in the production of a truncated but partially functional dystrophin protein. BMD patients, therefore, have milder symptoms, delayed disease progression, and longer life expectancy compared to DMD patients.1,2,3
Casimersen is an antisense phosphorodiamidate morpholino oligonucleotide designed to bind to exon 45 of the DMD pre-mRNA and prevent its inclusion within the mature mRNA before translation.4,7 It is estimated that around 8% of DMD patients may benefit from exon 45 skipping, in which the exclusion of this exon results in the production of an internally truncated and at least partly functional dystrophin protein.4,7,5 Although fibrotic or fatty muscle tissue developed previously cannot be improved, this therapy aims to slow further disease progression through the production of partially functional dystrophin and alleviation of the pathogenic mechanism of muscle tissue necrosis.1,2
| TARGET | ACTIONS | ORGANISM |
|---|---|---|
| ADMD gene (exon 45 casimersen target site) |
binder
|
Humans |
Absorption
DMD patients receiving IV doses of 4-30 mg/kg/week revealed exposure in proportion to dose with no accumulation of casimersen in plasma with once-weekly dosing. Following a single IV dose, casimersen Cmax was reached by the end of infusion. Inter-subject variability, as measured by the coefficient of variation, ranged from 12-34% for Cmax and 16-34% for AUC.7
Pre-clinical studies in nonhuman primates (cynomolgus monkeys) investigated the pharmacokinetics of once-weekly casimersen administered at doses of 5, 40, and 320 mg/kg. On days 1 and 78, the 5 mg/kg dose resulted in a Cmax of 19.5 ± 3.43 and 21.6 ± 5.60 μg/mL and an AUC0-t of 24.9 ± 5.17 and 26.9 ± 7.94 μg*hr/mL. The 40 mg/kg dose resulted in a Cmax of 208 ± 35.2 and 242 ± 71.1 μg/mL and an AUC0-t of 283 ± 68.5 and 320 ± 111 μg*hr/mL. Lastly, the 320 mg/kg dose resulted in a a Cmax of 1470 ± 88.1 and 1490 ± 221 μg/mL and an AUC0-t of 1960 ± 243 and 1930 ± 382 μg*hr/mL.4
Volume of distribution
Casimersen administered at 30 mg/kg had a mean steady-state volume of distribution (%CV) of 367 mL/kg (28.9%).7
Protein binding
Casimersen binding to human plasma proteins is not concentration-dependent, ranging from 8.4-31.6%.7
Metabolism
Casimersen incubated with human hepatic microsomal preparations is metabolically stables and no metabolites are detected in plasma or urine.7
Route of elimination
Casimersen is predominantly (more than 90%) excreted in the urine unchanged with negligible fecal excretion.7
Half-life
Casimersen has an elimination half-life of 3.5 ± 0.4 hours.7
Clearance
Casimersen administered at 30 mg/kg has a plasma clearance of 180 mL/hr/kg.7
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Amondys 45 | Injection | 50 mg/1mL | Intravenous | Sarepta Therapeutics, Inc. | 2021-02-25 | Not applicable |  |
Synthesis Reference
Diane Elizabeth Frank and Richard K. Bestwick, “Exon skipping oligomers for muscular dystrophy.” U.S. Patent US20190262375A1, issued August 29, 2019.
PATENT
https://patents.google.com/patent/WO2017205879A2/en
also
WO 2021025899
References
- ^ Jump up to:a b c d e “Amondys 45- casimersen injection”. DailyMed. Retrieved 1 March 2021.
- ^ Jump up to:a b c d e f g h i j “FDA Approves Targeted Treatment for Rare Duchenne Muscular Dystrophy Mutation”. U.S. Food and Drug Administration (FDA) (Press release). 25 February 2021. Retrieved 25 February 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Sarepta Therapeutics Announces FDA Approval of Amondys 45 (casimersen) Injection for the Treatment of Duchenne Muscular Dystrophy (DMD) in Patients Amenable to Skipping Exon 45” (Press release). Sarepta Therapeutics. 25 February 2021. Retrieved 25 February 2021 – via GlobeNewswire.
- ^ Rodrigues M, Yokota T (2018). “An Overview of Recent Advances and Clinical Applications of Exon Skipping and Splice Modulation for Muscular Dystrophy and Various Genetic Diseases”. Exon Skipping and Inclusion Therapies. Methods in Molecular Biology. 1828. Clifton, N.J. pp. 31–55. doi:10.1007/978-1-4939-8651-4_2. ISBN 978-1-4939-8650-7. PMID 30171533.
- ^ “Casimersen Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 4 June 2019. Retrieved 25 February 2021.
General References
- Wein N, Alfano L, Flanigan KM: Genetics and emerging treatments for Duchenne and Becker muscular dystrophy. Pediatr Clin North Am. 2015 Jun;62(3):723-42. doi: 10.1016/j.pcl.2015.03.008. Epub 2015 Apr 20. [PubMed:26022172]
- Verhaart IEC, Aartsma-Rus A: Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol. 2019 Jul;15(7):373-386. doi: 10.1038/s41582-019-0203-3. [PubMed:31147635]
- Mercuri E, Bonnemann CG, Muntoni F: Muscular dystrophies. Lancet. 2019 Nov 30;394(10213):2025-2038. doi: 10.1016/S0140-6736(19)32910-1. [PubMed:31789220]
- Carver MP, Charleston JS, Shanks C, Zhang J, Mense M, Sharma AK, Kaur H, Sazani P: Toxicological Characterization of Exon Skipping Phosphorodiamidate Morpholino Oligomers (PMOs) in Non-human Primates. J Neuromuscul Dis. 2016 Aug 30;3(3):381-393. doi: 10.3233/JND-160157. [PubMed:27854228]
- Rodrigues M, Yokota T: An Overview of Recent Advances and Clinical Applications of Exon Skipping and Splice Modulation for Muscular Dystrophy and Various Genetic Diseases. Methods Mol Biol. 2018;1828:31-55. doi: 10.1007/978-1-4939-8651-4_2. [PubMed:30171533]
- Smith CIE, Zain R: Therapeutic Oligonucleotides: State of the Art. Annu Rev Pharmacol Toxicol. 2019 Jan 6;59:605-630. doi: 10.1146/annurev-pharmtox-010818-021050. Epub 2018 Oct 9. [PubMed:30285540]
- FDA Approved Drug Products: AMONDYS 45 (casimersen) injection [Link]
External links
- “Casimersen”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02500381 for “Study of SRP-4045 and SRP-4053 in DMD Patients (ESSENCE)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Amondys 45 |
| Other names | SRP-4045 |
| License data |
|
| Routes of administration |
Intravenous |
| Drug class | Antisense oligonucleotide |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| DrugBank | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C268H424N124O95P22 |
| Molar mass | 7584.536 g·mol−1 |
////////////Casimersen, FDA 2021, APPROVALS 2021, カシメルセン , Exon-45: NG-12-0064, SRP-4045, WHO 10354, Amondys 45, Antisense oligonucleotide, Duchenne muscular dystrophy
#Casimersen, #FDA 2021, #APPROVALS 2021, #カシメルセン , #Exon-45: NG-12-0064, #SRP-4045, #WHO 10354, #Amondys 45, #Antisense oligonucleotide, #Duchenne muscular dystrophy
Sequence:
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| modified base | c-1 | 5′-ester |
| modified base | c-1 | modified cytidine |
| modified base | a-2 | modified adenosine |
| modified base | a-3 | modified adenosine |
| modified base | u-4 | m5u |
| modified base | u-4 | modified uridine |
| modified base | g-5 | modified guanosine |
| modified base | c-6 | modified cytidine |
| modified base | c-7 | modified cytidine |
| modified base | a-8 | modified adenosine |
| modified base | u-9 | modified uridine |
| modified base | u-9 | m5u |
| modified base | c-10 | modified cytidine |
| modified base | c-11 | modified cytidine |
| modified base | u-12 | m5u |
| modified base | u-12 | modified uridine |
| modified base | g-13 | modified guanosine |
| modified base | g-14 | modified guanosine |
| modified base | a-15 | modified adenosine |
| modified base | g-16 | modified guanosine |
| modified base | u-17 | modified uridine |
| modified base | u-17 | m5u |
| modified base | u-18 | modified uridine |
| modified base | u-18 | m5u |
| modified base | c-19 | modified cytidine |
| modified base | c-20 | modified cytidine |
| modified base | u-21 | m5u |
| modified base | u-21 | modified uridine |
| modified base | g-22 | modified guanosine |
| uncommon link | c-1 – a-2 | unavailable |
| uncommon link | a-2 – a-3 | unavailable |
| uncommon link | a-3 – u-4 | unavailable |
| uncommon link | u-4 – g-5 | unavailable |
| uncommon link | g-5 – c-6 | unavailable |
| uncommon link | c-6 – c-7 | unavailable |
| uncommon link | c-7 – a-8 | unavailable |
| uncommon link | a-8 – u-9 | unavailable |
| uncommon link | u-9 – c-10 | unavailable |
| uncommon link | c-10 – c-11 | unavailable |
| uncommon link | c-11 – u-12 | unavailable |
| uncommon link | u-12 – g-13 | unavailable |
| uncommon link | g-13 – g-14 | unavailable |
| uncommon link | g-14 – a-15 | unavailable |
| uncommon link | a-15 – g-16 | unavailable |
| uncommon link | g-16 – u-17 | unavailable |
| uncommon link | u-17 – u-18 | unavailable |
| uncommon link | u-18 – c-19 | unavailable |
| uncommon link | c-19 – c-20 | unavailable |
| uncommon link | c-20 – u-21 | unavailable |
| uncommon link | u-21 – g-22 | unavailable |