
















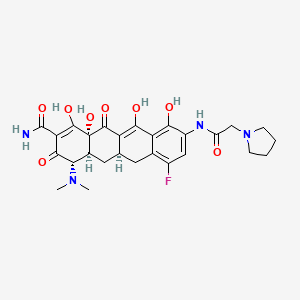
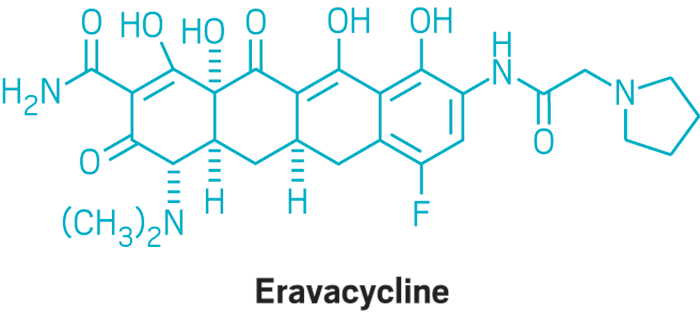
Eravacycline
http://www.ama-assn.org/resources/doc/usan/eravacycline.pdf
1-Pyrrolidineacetamide, N-[(5aR,6aS,7S,10aS)-9-(aminocarbonyl)-7-(dimethylamino)-
4-fluoro-5,5a,6,6a,7,10,10a,12-octahydro-1,8,10a,11-tetrahydroxy-10,12-dioxo-2-
naphthacenyl]-
(4S,4aS,5aR,12aS)-4-(dimethylamino)-7-fluoro-3,10,12,12a-tetrahydroxy-1,11-dioxo-9-
[(pyrrolidin-1-ylacetyl)amino]-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide
MOLECULAR FORMULA C27H31FN4O8
MOLECULAR WEIGHT 558.6
SPONSOR Tetraphase Pharmaceuticals, Inc.
CODE DESIGNATION TP-434
CAS REGISTRY NUMBER 1207283-85-9
WHO NUMBER 9702
Eravacycline (TP-434) is a synthetic fluorocycline antibiotic in development by Tetraphase Pharmaceuticals. It is closely related to the glycylglycine antibiotic tigecycline and the tetracycline class of antibiotics. It has a broad spectrum of activity including many multi-drug resistant strains of bacteria. Phase III studies in complicated intra-abdominal infections (cIAI) [1] and complicated urinary tract infections (cUTI)[2] were recently completed with mixed results. Eravacylcine has been designated as a Qualified Infectious Disease Product (QIDP), as well as for fast track approval by the FDA.[3]
| Inventors | Jingye Zhou, Xiao-Yi Xiao, Louis Plamondon, Diana Katharine Hunt, Roger B. Clark, Robert B. Zahler, |
| Applicant | Tetraphase Pharmaceuticals, Inc. |
Example 1. Synthesis of Compounds of Structural Formula (I).
The compounds of the invention can be prepared according the synthetic scheme shown in Scheme 1.

Compound 34

1H NMR (400 MHz, CD3OD) δ 8 22 (d, J= 1 1.0 Hz, 1 H), 4.33 (s, 2H), 4.10 (S3 1H), 3 83-3.72 (m, 2H), 3.25-2.89 (m, 12H), 2.32-2.00 (m, 6H), 1.69-1.56 (m, 1H); MS (ESI) m/z 559.39 (M+H).
Medical Uses
Eravacycline has shown broad spectrum of activity against a variety of Gram-positive and Gram-negative bacteria, including multi-drug resistant strains, such as methicillin-resistant Staphylococcus aureus (MRSA) and carbapenem-resistant Enterobacteriaceae.[4] It is currently being formulated as for intravenous and oral administration.

PATENT
Eravacylme is a tetracycline antibiotic that has demonstrated broad spectrum activity against a wide variety of multi-drug resistant Gram-negative, Gram-positive and anaerobic bacteria in humans. In Phase I and Phase II clinical trials, eravacycline also demonstrated a favorable safety and tolerability profile. In view of its attractive
pharmacological profile, synthetic routes to eravacycline and, in particular, synthetic routes that result in suitable quantities of eravacycline for drag development and manufacturing, are becoming increasingly important.
As described in International Publication No . WO 2010/017470, eravacycline is conveniently synthesized from 7-fluorosancycline, another tetracycline. 7-Fluorosancycline can be synthesized, in turn, from commercially available 7-ammosancycline or a protected derivative thereof. However, very few procedures for the conversion of (^-ammo-substituted tetracyclines, such as 7-aminosancycline, to C7-fiuoro-substituted tetracyclines, such as 7-fluorosancycline, have been reported, and those that have are not suitable to be deployed at production-scale.
Therefore, there is a need for improved processes, particularly improved production -scale processes, for converting C7-amino-substituted tetracyclines to C7-fluoro-substituted tetracyclines.

Example 3. Preparation of Eravacycline From 9-Aminosancycline Using a Photolytic Fluorination

[00158] Sancycline (0.414 g, 1.0 mmol) was dissolved in trifiuoroacetic acid (TFA). The solution was cooled to 0 °C. To the solution was added N-bromosuccinimide (NBS, 0.356 g, 2.1 mmol). The reaction was complete after stirring at 0 °C for 1 h. The reaction mixture was allowed to warm to rt. Solid NO3 (0.1 Ig, 0.11 mrnoi) was added and the reaction mixture was stirred at rt for 1 h. The reaction solution was added to 75 mL cold diethyl ether. The precipitate was collected by filtration and dried to give 0.46 g of compound 6. Compound 6 can then be reduced to compounds 7, 8, or 9 using standard procedures.



13
[00159] 9-Aminosancycline (7, 1 g, 0233 mmol) was dissolved in 20 mL sulfuric acid and the reaction was cooled using an ice bath. Potassium nitrate (235 mg, 0.233 mmol) was added in several portions. After stirring for 15 min, the reaction mixture was added to 400 mL MTBE followed by cooling using an ice bath. The solid was collected by filtration. The filter cake was dissolved in 10 mL water and the pH of the aqueous solution was adjusted to 5.3 using 25% aqueous NaOH. The resulting suspension was filtered, and the filter cake was dried to give 1 g compound 10: MS (ESI) m/z 475.1 (M+l).
[00160] Compound 10 (1.1 g) was dissolved in 20 mL of water and 10 mL of acetonitrile. To the solution was added acyl chloride 3 (in two portions: 600 mg and 650 mg). The pH of the reaction mixture was adjusted to 3.5 using 25% aqueous NaOH. Another portion of acyl chloride (800 mg) was added. The reaction was monitored by HPLC analysis. Product 11 was isolated from the reaction mixture by preparative HPLC. Lyophilization gave 1.1 g of compound 11: MS (ESI) m/z 586.3 (M+l).
[00161] Compound 11 (1.1 g) was dissolved in methanol. To the solution was added concentrated HC1 (0.5 mL) and 10% Pd-C (600 mg). The reaction mixture was stirred under a hydrogen atmosphere (balloon). After the reaction was completed, the catalyst was removed by filtration. The filtrate was concentrated to give 1 g of compound 12: ‘H NMR (400 MHz, DMSO), 8.37 (s, 1H), 4.38-4.33 (m, 3H), 3.70 (br s, 2H), 3.30-2.60 (m, 1211), 2.36-2.12 (m, 2H), 2.05-1.80 (m, 4H), 1.50-1.35 (m, 1H); MS (ESI) m/z 556.3 (M+l).
[00162] Compound 12 (150 mg) was dissolved in 1 mL of 48% HBF4. To the solution was added 21 mg of NaN02. After compound 12 was completely converted to compound 13 (LC/MS m/z 539.2), the reaction mixture was irradiated with 254 nm light for 6 h while being cooled with running water. The reaction mixture was purified by preparative HPLC using acetonitrile and 0.05 N aqueous HCl as mobile phases to yield the compound 4 (eravacyclme, 33 mg) as a bis-HCl salt (containing 78% of 4 and 10% of the 7-H byproduct, by HPLC): MS (ESI) m/z 559.3 (M+l).
PAPER
Exploring the Boundaries of “Practical”: De Novo Syntheses of Complex Natural Product-Based Drug Candidates
PAPER
Journal of Medicinal Chemistry (2012), 55(2), 597-605
Fluorocyclines. 1. 7-Fluoro-9-pyrrolidinoacetamido-6-demethyl-6-deoxytetracycline: A Potent, Broad Spectrum Antibacterial Agent

This and the accompanying report (DOI: 10.1021/jm201467r) describe the design, synthesis, and evaluation of a new generation of tetracycline antibacterial agents, 7-fluoro-9-substituted-6-demethyl-6-deoxytetracyclines (“fluorocyclines”), accessible through a recently developed total synthesis approach. These fluorocyclines possess potent antibacterial activities against multidrug resistant (MDR) Gram-positive and Gram-negative pathogens. One of the fluorocyclines, 7-fluoro-9-pyrrolidinoacetamido-6-demethyl-6-deoxytetracycline (17j, also known as TP–434, 50th Interscience Conference on Antimicrobial Agents and Chemotherapy Conference, Boston, MA, September 12–15, 2010, poster F1–2157), is currently undergoing phase 2 clinical trials in patients with complicated intra-abdominal infections (cIAI).
(4S,4aS,5aR,12aS)-4-(Dimethylamino)-7-fluoro-3,10,12,12a-tetrahydroxy-1,11-dioxo-9-[2-(pyrrolidin-1-yl)acetamido]-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide (17j)
PAPER
Journal of Organic Chemistry (2017), 82(2), 936-943
A Divergent Route to Eravacycline

Abstract

A convergent route to eravacycline (1) has been developed by employing Michael–Dieckmann cyclization between enone 3 and a fully built and protected left-hand piece (LHP, 2). After construction of the core eravacycline structure, a deprotection reaction was developed, allowing for the isoxazole ring opening and global deprotection to be achieved in one pot. The LHP is synthesized from readily available 4-fluoro-3-methylphenol in six steps featuring a palladium-catalyzed phenyl carboxylation in the last step.
Eravacycline di-HCl salt (1) as a yellow solid: purity 97.9%; mp 197–199 °C dec. The spectral data matched those from original sample as reported in our previous publication.(3)
3 Ronn, M.; Zhu, Z.; Hogan, P. C.; Zhang, W.-Y.; Niu, J.; Katz, C. E.; Dunwoody, N.; Gilicky, O.; Deng, Y.;Hunt, D. K.; He, M.; Chen, C.-L.; Sun, C.; Clark, R. B.; Xiao, X.-Y. Org. Process Res. Dev. 2013, 17, 838–845, DOI: 10.1021/op4000219

PAPER
WO 2016065290
PAPER
Organic Process Research & Development (2013), 17(5), 838-845

Process research and development of the first fully synthetic broad spectrum 7-fluorotetracycline in clinical development is described. The process utilizes two key intermediates in a convergent approach. The key transformation is a Michael–Dieckmann reaction between a suitable substituted aromatic moiety and a key cyclohexenone derivative. Subsequent deprotection and acylation provide the desired active pharmaceutical ingredient in good overall yield.
Free base of 7. HPLC (248 nm) showed 97.1% purity with 0.80% of the corresponding impurity from 19, 1.2% of epimer of 7, and 0.80% the corresponding impurity from 20 (102g, 88.7% yield) product as a yellow solid.
1H NMR (CDCl3, 400 MHz, δ): 9.81 (d, 1H), 3.29 (s, 2H), 3.04 (m, 3H); 2.68 (bs, 4H), 2.62 (m, 1H), 2.44 (bs, 6H), 2.23 (t, 1H), 1.99 (s, 1H), 1.84 (bs, 4H), 1.5 (bs, 1H).
MS (ES) m/z calcd for +H: 559.2 (100.0), 560.2 (29.9), 561.2 (5.9); found: 559.3, 560.2, 561.3.
Give 7·2HCl (531 g containing 6.9% by weight water, ∼ 784 mmol). HPLC (248 nm) indicated a 96.6% purity with 0.64% of the corresponding impurity from 19, 1.3% of epimer of 7, and 1.3% the corresponding impurity from 20.
1H NMR, (d6-DMSO, 400 MHz, δ): 11.85 (s, 1H), 10.28 (s, 1H), 9.56 (s, 1H); 8.99 (s, 1H), 8.04 (d, J = 10.95 Hz, 1H), 4.32 (m, 1H), 4.30 (s, 1H), 3.58 (bs, 2H), 3.09 (bs, 2H), 3.007 (m, 1H); 2.94 (m, 2H), 2.8 (s, 6H), 2.15–2.32 (m, 2H), 1.92 (bd, 4H), 1.44 (m, 1H).
13C NMR (d6-DMSO, 100 MHz)193.74, 191.84, 187.45, 175.72, 171.88, 164.45, 152.12, 151.26, 148.93, 148.86, 125.13, 125.03, 122.79, 122.60, 116.00, 115.72, 115.25, 108.12, 95.38, 74.06, 67.78, 55.47, 53.91, 34.95, 33.98, 31.41, 26.45, 22.9.
MS (ES) m/zcalcd for +H: 559.2 (100.0), 560.2 (29.9), 561.2 (5.9); found: 559.3, 560.2, 561.3.
PAPER
Natural product synthesis in the age of scalability – Natural …
RSC Publishing – Royal Society of Chemistry
10.1039/C3NP70090A
Natural product synthesis in the age of scalability
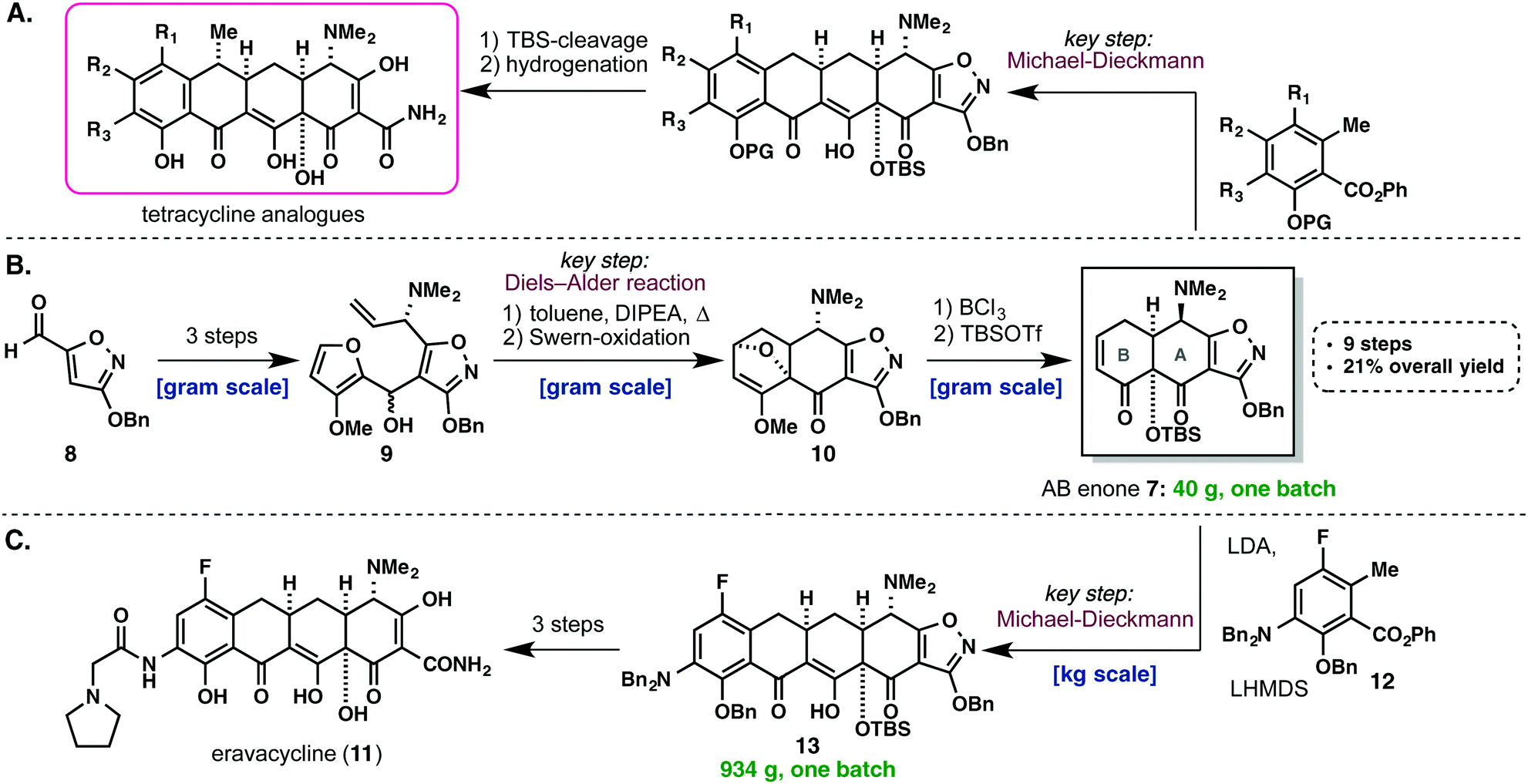
Tetracycline (Myers/Tetraphase, 2005–2013). Myers’ convergent approach to the tetracyclines is a great example of how a scalable synthesis of a key intermediate en route to a natural product can fuel the discovery of entirely new drug candidates. These broad-spectrum polyketide antibiotics have been widely used in human and veterinary medicine, but, due to the development of tetracycline-resistant strains, there is an unmet need for novel tetracycline drugs. Pioneering work in this eld has been achieved by the Myers’ group, who published a landmark synthetic approach to the tetracycline class of antibiotics in 2005.16 Using this route, over 3,000 fully synthetic tetracyclines have been prepared to date. Central to their strategy was the synthesis of a highly versatile intermediate, AB enone 7, 17 which enabled the convergent construction of novel tetracycline antibiotics (Scheme 3, A).18 Naturally, the route to 7 had to be practical and amenable to large-scale synthesis and consequently, the synthetic approaches to this building block have become more and more practical and efficient with every new generation. In 2007, Myers published their rst practical and enantioselective approach to 7 (Scheme 3, B).17 The route started from 8, which can be accessed in multi-hundred gram amounts from commercially available 3-hydroxy-5-isoxazolecarboxylate (not shown) by O-benzylation followed by DIBAL reduction. In a three-step sequence, 8 was transformed into carbinol 9. In the key step of the sequence, 9 underwent an intramolecular Diels–Alder reaction to give a mixture of 4 diastereomeric cycloadducts, which, aer Swern oxidation, could be readily separated by ash column chromatography to afford 10. Finally, boron trichloride mediated opening of the oxabicyclic ring system and demethylation, followed by TBS protection of the tertiary hydroxyl-group, afforded 40 g of the AB enone 7 in 21% overall yield, over nine steps from commercial material. Slight modications of this route have allowed for the preparation of >20 kg batches of the AB enone. The availability of large-scale batches of 7 has both enabled the discovery and the development of eravacycline (11), the rst fully synthetic tetracycline analog in clinical development, from Tetraphase Pharmaceuticals. In their process route,19 the key Michael– Dieckmann cyclization between 7 and 12 was carried out on kg-scale and afforded 13 in 93.5% yield. This compound was transformed into eravacycline (11) in 3 more steps, including TBS-cleavage, hydrogenolysis and amide bond formation. Using this process, several kg of 11 have been prepared to date to support clinical studies. Finally, a third- and fourth-generation route to 7 has recently been published by Myers that is not only shorter than previous routes, but also amenable of structural modications of the AB-ring enone.20
16 M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, Science, 2005, 308, 395–398. 17 J. D. Brubaker and A. G. Myers, Org. Lett., 2007, 9, 3523–3525. 18 (a) C. Sun, Q. Wang, J. D. Brubaker, P. M. Wright, C. D. Lerner, K. Noson, M. Charest, D. R. Siegel, Y.-M. Wang and A. G. Myers, J. Am. Chem. Soc., 2008, 130, 17913–17927; (b) X.-Y. Xiao, D. K. Hunt, J. Zhou, R. B. Clark, N. Dunwoody, C. Fyfe, T. H. Grossman, W. J. O’Brien, L. Plamondon, M. R¨onn, C. Sun, W.-Y. Zhang and J. A. Sutcliffe, J. Med. Chem., 2012, 55, 597–605; (c) R. B. Clark, M. He, C. Fyfe, D. Loand, W. J. O’Brien, L. Plamondon, J. A. Sutcliffe and X.-Y. Xiao, J. Med. Chem., 2011, 54, 1511–1528; (d) R. B. Clark, D. K. Hunt, M. He, C. Achorn, C.-L. Chen, Y. Deng, C. Fyfe, T. H. Grossman, P. C. Hogan, W. J. O’Brien, L. Plamondon, M. R¨onn, J. A. Sutcliffe, Z. Zhu and X.-Y. Xiao, J. Med. Chem., 2012, 55, 606–622; (e) C. Sun, D. K. Hunt, R. B. Clark, D. Loand, W. J. O’Brien, L. Plamondon and X.-Y. Xiao, J. Med. Chem., 2011, 54, 3704–3731. 19 M. Ronn, Z. Zhu, P. C. Hogan, W.-Y. Zhang, J. Niu, C. E. Katz, N. Dunwoody, O. Gilicky, Y. Deng, D. K. Hunt, M. He, C.-L. Chen, C. Sun, R. B. Clark and X.-Y. Xiao, Org. Process Res. Dev., 2013, 17, 838–845. 20 D. A. Kummer, D. Li, A. Dion and A. G. Myers, Chem. Sci., 2011, 2, 1710–1718. 21 (a) G. R. Pettit, Z. A. Chicacz, F. Gao, C. L. Herald, M. R. Boyd, J. M. Schmidt and J. N. A. Hooper, J. Org. Chem., 1993, 58, 1302–1304; (b) M. Kobayashi, S. Aoki, H. Sakai, K. Kawazoe, N. Kihara, T. Sasaki and I. Kitagawa, Tetrahedron Lett., 1993, 34, 2795–2798. 22 (a) J. Guo, K. J. Duffy, K. L. Stevens, P. I. Dalko, R. M. Roth, M. M. Hayward and Y. Kishi, Angew. Chem., Int. Ed., 1998, 37, 187–190; (b) M. M. Hayward, R. M. Roth, K. J. Duffy, P. I. Dalko, K. L. Stevens, J. Guo and Y. Kishi, Angew. Chem., Int. Ed., 1998, 37, 190–196; (c) I. Paterson, D. Y. K. Chen, M. J. Coster, J. L. Acena, J. Bach, ˜ K. R. Gibson, L. E. Keown, R. M. Oballa, T. Trieselmann, D. J. Wallace, A. P. Hodgson and R. D. Norcross, Angew. Chem., Int. Ed., 2001, 40, 4055–4060; (d) M. T. Crimmins, J. D. Katz, D. G. Washburn, S. P. Allwein and L. F. McAtee, J. Am. Chem. Soc., 2002, 124, 5661–5663; (e) M. Ball, M. J. Gaunt, D. F. Hook, A. S. Jessiman, S. Kawahara, P. Orsini, A. Scolaro, A. C. Talbot, H. R. Tanner, S. Yamanoi and S. V. Ley, Angew. Chem., Int. Ed., 2005, 44, 5433–5438. 23 A. B. Smith, T. Tomioka, C. A. Risatti, J. B. Sperry and C. Sfouggatakis, Org. Lett., 2008, 10, 4359–4362. 24 (a) U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed. Engl., 1971, 10, 496–497; (b) Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615–1621; (c) B. List, Tetrahedron, 2002, 58, 5573–5590; (d) A. B. Northrup and D. W. C. MacMillan, Science, 2004, 305, 1752–1755; (e) A. B. Northrup, I. K. Mangion, F. Hettche and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2004, 43, 2152– 2154. 25 A. B. Smith, C. Sfouggatakis, D. B. Gotchev, S. Shirakami, D. Bauer, W. Zhu and V. A. Doughty, Org. Lett., 2004, 6, 3637–3640. 26 A. B. Smith, C. A. Risatti, O. Atasoylu, C. S. Bennett, J. Liu, H. Cheng, K. TenDyke and Q. Xu, J. Am. Chem. Soc., 2011, 133, 14042–14053. 27 A. R. Carroll, E. Hyde, J. Smith, R. J. Quinn, G. Guymer and P. I. Forster, J. Org. Chem., 2005, 70, 1096–1099. 2W. J. O’Brien, L. Plamondon, M. R¨onn, C. Sun, W.-Y. Zhang and J. A. Sutcliffe, J. Med. Chem., 2012, 55, 597–605; (c) R. B. Clark, M. He, C. Fyfe, D. Loand, W. J. O’Brien, L. Plamondon, J. A. Sutcliffe and X.-Y. Xiao, J. Med. Chem., 2011, 54, 1511–1528; (d) R. B. Clark, D. K. Hunt, M. He, C. Achorn, C.-L. Chen, Y. Deng, C. Fyfe, T. H. Grossman, P. C. Hogan, W. J. O’Brien, L. Plamondon, M. R¨onn, J. A. Sutcliffe, Z. Zhu and X.-Y. Xiao, J. Med. Chem., 2012, 55, 606–622; (e) C. Sun, D. K. Hunt, R. B. Clark, D. Loand, W. J. O’Brien, L. Plamondon and X.-Y. Xiao, J. Med. Chem., 2011, 54, 3704–3731. 19 M. Ronn, Z. Zhu, P. C. Hogan, W.-Y. Zhang, J. Niu, C. E. Katz, N. Dunwoody, O. Gilicky, Y. Deng, D. K. Hunt, M. He, C.-L. Chen, C. Sun, R. B. Clark and X.-Y. Xiao, Org. Process Res. Dev., 2013, 17, 838–845. 20 D. A. Kummer, D. Li,
PAPER
Applications of biocatalytic arene ipso,ortho cis-dihydroxylation in synthesis
10.1039/C3CC49694E
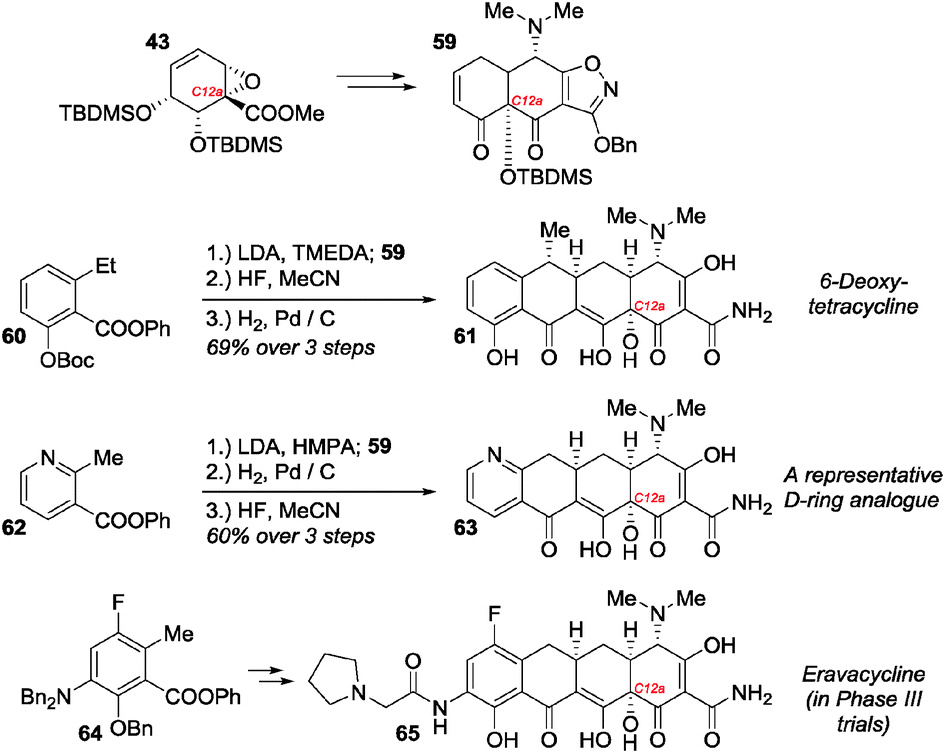
In 2005, Myers and co-workers reported the first use of 4 in complex natural product total synthesis.13 From their previously reported building block 43, tricyclic diketone 59 was accessible in a further 7 steps (10% overall yield from benzoate, Scheme 7). Diketone 59 serves as a common precursor to the tetracycline AB-ring system and may be coupled with D-ring precursors such as 60 by a Michael–Dieckmann cascade cyclisation that forms the C-ring. Thus, after deprotection, the natural product ()-6-deoxytetracycline 61 is accessible in 14 steps and 7.0% overall yield from benzoate. Several points about the synthesis are noteworthy. The yield represents an improvement of orders of magnitude over the yields for all previously reported total syntheses of tetracyclines. Thus, for the first time, novel tetracycline analogues became accessible in useful quantities; union of 62 with 59 to access 63 is a representative example. Secondly, previous total syntheses of tetracyclines had been bedevilled by the difficulty of installing the C12a tertiary alcohol at a late stage.14c The Myers approach is conceptually distinct in that the C12a hydroxyl group is installed in the very first step, i.e. it is the hydroxyl group deriving from the microbial ipso hydroxylation. Finally, apart from the C12a stereocentre, all other stereocentres in the final tetracyclines are set under substrate control. Thus, all the stereochemical information in the final products may be considered ultimately to be of enzymatic origin. In the years following the Myers group’s initial disclosure, the methodology has been extended and improved to allow for the preparation of a greater diversity of novel tetracycline analogues.14 This has culminated in the development of eravacycline 65 (accessed from 59 and 64) by Tetraphase Pharmaceuticals.15 Eravacycline is indicated for treatment of multidrug-resistant infections and is currently in phase III trials.
13 (a) M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, Science, 2005, 308, 395; (b) M. G. Charest, D. R. Siegel and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 8292.
14 (a) J. D. Brubaker and A. G. Myers, Org. Lett., 2007, 9, 3523; (b) C. Sun, Q. Wang, J. D. Brubaker, P. M. Wright, C. D. Lerner, K. Noson, M. Charest, D. R. Siegel, Y.-M. Wang and A. G. Myers, J. Am. Chem. Soc., 2008, 130, 17913; (c) D. A. Kummer, D. Li, A. Dion and A. G. Myers, Chem. Sci., 2011, 2, 1710; (d) P. M. Wright and A. G. Myers, Tetrahedron, 2011, 67, 9853.
15 (a) R. B. Clark, M. He, C. Fyfe, D. Lofland, W. J. O’Brien, L. Plamondon, J. A. Sutcliffe and X.-Y. Xiao, J. Med. Chem., 2011, 54, 1511; (b) C. Sun, D. K. Hunt, R. B. Clark, D. Lofland, W. J. O’Brien, L. Plamondon and X.-Y. Xiao, J. Med. Chem., 2011, 54, 3704; (c) X.-Y. Xiao, D. K. Hunt, J. Zhou, R. B. Clark, N. Dunwoody, C. Fyfe, T. H. Grossman, W. J. O’Brien, L. Plamondon, M. Ro¨nn, C. Sun, W.-Y. Zhang and J. A. Sutcliffe, J. Med. Chem., 2012, 55, 597; (d) R. B. Clark, D. K. Hunt, M. He, C. Achorn, C.-L. Chen, Y. Deng, C. Fyfe, T. H. Grossman, P. C. Hogan, W. J. O’Brien, L. Plamondon, M. Ro¨nn, J. A. Sutcliffe, Z. Zhu and X.-Y. Xiao, J. Med. Chem., 2012, 55, 606; (e) M. Ronn, Z. Zhu, P. C. Hogan, W.-Y. Zhang, J. Niu, C. E. Katz, N. Dunwoody, O. Gilicky, Y. Deng, D. K. Hunt, M. He, C.-L. Chen, C. Sun, R. B. Clark and X.-Y. Xiao, Org. Process Res. Dev., 2013, 17, 838; ( f ) R. B. Clark, M. He, Y. Deng, C. Sun, C.-L. Chen, D. K. Hunt, W. J. O’Brien, C. Fyfe, T. H. Grossman, J. A. Sutcliffe, C. Achorn, P. C. Hogan, C. E. Katz, J. Niu, W.-Y. Zhang, Z. Zhu, M. Ro¨nn and X.-Y. Xiao, J. Med. Chem., 2013, 56, 8112.
WO 2017125557, Crystalline forms of eravacycline dihydrochloride or its solvates or hydrates. Also claims a process for the preparation of an eravacycline intended for oral or parenteral use, for the treatment of bacterial infections, preferably intra-abdominal and urinary tract infections caused by multidrug resistant gram negative pathogens. Follows on from WO2017097891 .
Tetraphase Pharmaceuticals is developing eravacycline, a fully synthetic fluorocycline antibiotic and the lead from a series of tetracycline analogs which includes TP-221 and TP-170, for treating bacterial infections.
Eravacycline is a tetracycline antibiotic chemically designated (4S,4aS,5aR,l2aS)-4-(Dimethylamino)-7-fluoro-3 ,10,12,12a-tetrahydroxy- 1,11 -dioxo-9-[2-(-pyrrolidin- 1 -yl)acetamido]-l,4,4a,5,5a,6,l l ,12a-octahydrotetracene-2-carboxamide and can be represented by the following chemical structure according to formula (I).
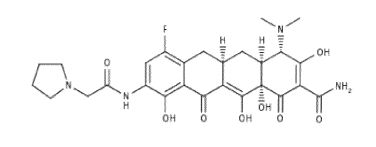
formula (I)
Eravacycline possesses antibacterial activity against Gram negative pathogens and Gram positive pathogens, in particular against multidrug resistant (MDR) Gram negative pathogens and is currently undergoing phase III clinical trials in patients suffering from complicated intraabdominal infections (cIAI) and urinary tract infections (cUTI).
WO 2010/017470 Al discloses eravacycline as compound 34. Eravacycline is described to be prepared according to a process, which is described in more detail only for related compounds.
The last step of this process involves column chromatography with diluted hydrochloric acid/ acetonitrile, followed by freeze drying.
WO 2012/021829 Al discloses pharmaceutically acceptable acid and base addition salts of eravacycline in general and a general process for preparing the same involving reacting eravacycline free base with the corresponding acids and bases, respectively. On page 15, lines 3 to 6, a lyophilized powder containing an eravacycline salt and mannitol is disclosed.
Xiao et. al. “Fluorocyclines. 1. 7-Fluoro-9-pyrrolidinoacetamido-6-demethyl-6-deoxytetracycline: A Potent, Broad Spectrum Antibacterial Agent” J. Med. Chem. 2012, 55, 597-605 synthesized eravacycline following the procedure for compounds 17e and 17i on page 603. After preparative reverse phase HPLC, compounds 17e and 17i were both obtained as bis-hydrochloride salts in form of yellow solids.
Ronn et al. “Process R&D of Eravacycline: The First Fully Synthetic Fluorocycline in Clinical Development” Org. Process Res. Dev. 2013, 17, 838-845 describe a process yielding eravacycline bis-hydrochloride as the final product. The last step involves precipitation of eravacycline bis-hydrochloride salt by adding ethyl acetate as an antisolvent to a solution of eravacycline bis-hydrochloride in an ethanol/ methanol mixture. The authors describe in some detail the difficulties during preparation of the bis-hydrochloride salt of eravacycline. According to Ronn et al. “partial addition of ethyl acetate led to a mixture containing suspended salt and a gummy form of the salt at the bottom of the reactor. At this stage, additional ethanol was added, and the mixture was aged with vigorous stirring until the gummy material also became a suspended solid.” In addition, after drying under vacuum the solid contained “higher than acceptable levels of ethanol”. “The ethanol was then displaced by water by placing a tray containing the solids obtained in a vacuum oven at reduced pressure (…) in the presence of an open vessel of water.” At the end eravacycline bis-hydrochloride salt containing about 4 to 6% residual moisture was obtained. The authors conclude that there is a need for additional improvements to the procedure along with an isolation step suitable for large scale manufacturing.
It is noteworthy that eravacycline or its salts are nowhere described as being a crystalline solid and that the preparation methods used for the preparation of eravacycline are processes like lyophilization, preparative column chromatography and precipitation, which typically yield amorphous material.
The cumbersome process of Ronn et al. points towards problems in obtaining eravacycline bis-hydrochloride in a suitable solid state, problems with scaleability of the available production process as well as problems with the isolation and drying steps of eravacycline.
In addition, amorphous solids can show low chemical stability, low physical stability, hygroscopicity, poor isolation and powder properties, etc.. Such properties are drawbacks for the use as an active pharmaceutical ingredients.
Thus, there is a need in pharmaceutical development for solid forms of an active pharmaceutical ingredient which demonstrate a favorable profile of relevant properties for formulation as a pharmaceutical composition, such as high chemical and physical stability, improved properties upon moisture contact, low(er) hygroscopicity and improved powder properties.
WO2010017470
WO 2017125557
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017125557&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
The invention relates to crystalline eravacycline bis-hydrochloride and to a process for its preparation. Furthermore, the invention relates to the use of crystalline eravacycline bis-hydrochloride for the preparation of pharmaceutical compositions. The invention further relates to pharmaceutical compositions comprising an effective amount of crystalline eravacycline bis-hydrochloride. The pharmaceutical compositions of the present invention can be used as medicaments, in particular for treatment and/ or prevention of bacterial infections e.g. caused by Gram negative pathogens or Gram positive pathogens, in particular caused by multidrug resistant Gram negative pathogens. The pharmaceutical compositions of the present invention can thus be used as medicaments for e.g. the treatment of complicated intra-abdominal and urinary tract infection
References
- ^ Jump up to:a b Solomkin, Joseph; Evans, David; Slepavicius, Algirdas; Lee, Patrick; Marsh, Andrew; Tsai, Larry; Sutcliffe, Joyce A.; Horn, Patrick (2016-11-16). “Assessing the Efficacy and Safety of Eravacycline vs Ertapenem in Complicated Intra-abdominal Infections in the Investigating Gram-Negative Infections Treated With Eravacycline (IGNITE 1) Trial: A Randomized Clinical Trial”. JAMA surgery. ISSN 2168-6262. PMID 27851857. doi:10.1001/jamasurg.2016.4237.
- ^ Jump up to:a b “Tetraphase Announces Top-Line Results From IGNITE2 Phase 3 Clinical Trial of Eravacycline in cUTI (NASDAQ:TTPH)”. ir.tphase.com. Retrieved 2016-11-20.
- Jump up^ “FDA Grants QIDP Designation to Eravacycline, Tetraphase’s Lead Antibiotic Product Candidate | Business Wire”. www.businesswire.com. Retrieved 2016-11-20.
- ^ Jump up to:a b Zhanel, George G.; Cheung, Doris; Adam, Heather; Zelenitsky, Sheryl; Golden, Alyssa; Schweizer, Frank; Gorityala, Bala; Lagacé-Wiens, Philippe R. S.; Walkty, Andrew (2016-04-01). “Review of Eravacycline, a Novel Fluorocycline Antibacterial Agent”. Drugs. 76 (5): 567–588. ISSN 1179-1950. PMID 26863149. doi:10.1007/s40265-016-0545-8.
- Jump up^ Sutcliffe, J. A.; O’Brien, W.; Fyfe, C.; Grossman, T. H. (2013-11-01). “Antibacterial activity of eravacycline (TP-434), a novel fluorocycline, against hospital and community pathogens”. Antimicrobial Agents and Chemotherapy. 57 (11): 5548–5558. ISSN 1098-6596. PMC 3811277
 . PMID 23979750. doi:10.1128/AAC.01288-13.
. PMID 23979750. doi:10.1128/AAC.01288-13. - Jump up^ Solomkin, Joseph S.; Ramesh, Mayakonda Krishnamurthy; Cesnauskas, Gintaras; Novikovs, Nikolajs; Stefanova, Penka; Sutcliffe, Joyce A.; Walpole, Susannah M.; Horn, Patrick T. (2014-01-01). “Phase 2, randomized, double-blind study of the efficacy and safety of two dose regimens of eravacycline versus ertapenem for adult community-acquired complicated intra-abdominal infections”. Antimicrobial Agents and Chemotherapy. 58 (4): 1847–1854. ISSN 1098-6596. PMC 4023720 . PMID 24342651. doi:10.1128/AAC.01614-13.
- Jump up^ Abdallah, Marie; Olafisoye, Olawole; Cortes, Christopher; Urban, Carl; Landman, David; Quale, John (2015-03-01). “Activity of eravacycline against Enterobacteriaceae and Acinetobacter baumannii, including multidrug-resistant isolates, from New York City”. Antimicrobial Agents and Chemotherapy. 59 (3): 1802–1805. ISSN 1098-6596. PMC 4325809 . PMID 25534744. doi:10.1128/AAC.04809-14.
- Jump up^ Fyfe, Corey; LeBlanc, Gabrielle; Close, Brianna; Nordmann, Patrice; Dumas, Jacques; Grossman, Trudy H. (2016-08-22). “Eravacycline is active against bacterial isolates expressing the polymyxin resistance gene mcr-1”. Antimicrobial Agents and Chemotherapy. 60: 6989–6990. ISSN 0066-4804. PMC 5075126 . PMID 27550359. doi:10.1128/AAC.01646-16.
- Jump up^ “http://www.healio.com/infectious-disease/antimicrobials/news/online/%7B3b5e5b8a-a5eb-4739-a402-3c88c22621d4%7D/phase-3-ignite4-trial-to-examine-safety-efficacy-of-iv-eravacycline-in-ciais”. www.healio.com. Retrieved 2016-11-20. External link in
|title=(help) - ^ Jump up to:a b “Tetraphase Pharmaceuticals Provides Update on Eravacycline Regulatory and Development Status (NASDAQ:TTPH)”. ir.tphase.com. Retrieved 2016-11-20.
- ^ Jump up to:a b c “Tetraphase Announces Positive Top-Line Results from Phase 3 IGNITE4 Clinical Trial in Complicated Intra-Abdominal Infections (NASDAQ:TTPH)”. ir.tphase.com. Retrieved 2017-07-27.
- ^ Jump up to:a b “Efficacy and Safety Study of Eravacycline Compared With Meropenem in Complicated Intra-abdominal Infections – Full Text View – ClinicalTrials.gov”. www.clinicaltrials.gov. Retrieved 2017-07-27.
- Jump up^ “http://www.healio.com/infectious-disease/antimicrobials/news/online/%7B8b0a64f5-6a4c-4b88-b5ac-9c1fe100778c%7D/ignite2-eravacycline-inferior-to-levofloxacin-but-iv-formulation-shows-promise”. www.healio.com. Retrieved 2016-11-20. External link in
|title=(help) - ^ Jump up to:a b “Efficacy and Safety Study of Eravacycline Compared With Ertapenem in Participants With Complicated Urinary Tract Infections – Full Text View – ClinicalTrials.gov”. www.clinicaltrials.gov. Retrieved 2017-07-27.
- ^ Jump up to:a b “Tetraphase Pharmaceuticals Doses First Patient in IGNITE3 Phase 3 Clinical Trial of Once-daily IV Eravacycline in cUTI (NASDAQ:TTPH)”. ir.tphase.com. Retrieved 2017-07-27.
- Jump up^ “Tetraphase reports 3Q loss”. Retrieved 2016-11-20.
- Jump up^ Feroldi, Brian (2016-11-20). “Why Tetraphase Pharmaceuticals Dropped 74% of Its Value in 2015 — The Motley Fool”. The Motley Fool. Retrieved 2016-11-20.
External links
- “Tetraphase Announces Top-Line Results From IGNITE2 Phase 3 Clinical Trial of Eravacycline in cUTI: Eravacycline Did Not Achieve Primary Endpoint in Pivotal Portion of cUTI Trial”
- Tetraphase pipeline
Notes
 |
|
| Names | |
|---|---|
| IUPAC name
(4S,4aS,5aR,12aS)-4-(Dimethylamino)-7-fluoro-3,10,12,12a-tetrahydroxy-1,11-dioxo-9-[(1-pyrrolidinylacetyl)amino]-1,4,4a,5,5a,6,11,12a-octahydro-2-tetracenecarboxamide
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
| KEGG | |
|
PubChem CID
|
|
| Properties | |
| C27H31FN4O8 | |
| Molar mass | 558.555 |
///////////////////////
OLD DATA FROM PREVIOUS BLOG POST
Tetraphase Pharmaceuticals Inc. (NASDAQ:TTPH) today announced that it will present two posters at IDWeek 2013 that examine the potential of its lead antibiotic candidate eravacycline to treat serious multi-drug resistant (MDR) infections. The first will highlight positive results of a Phase 1 study assessing the bronchopulmonary disposition safety and tolerability of eravacycline in healthy men and women; this study represents the first clinical assessment of eravacycline for potential use in treating pneumonia. The second poster will provide the results of a study that examined the activity of eravacycline in vitro against multiple Gram-negative and Gram-positive pathogens to set quality-control limits for monitoring eravacycline activity in future testing programs.
http://www.pharmiweb.com/pressreleases/pressrel.asp?ROW_ID=79335#.Uk6MOoanonU
More: http://www.pharmiweb.com/pressreleases/pressrel.asp?ROW_ID=79335#.Uk6MOoanonU#ixzz2gkEMTtQm
Eravacycline (TP-434) is a synthetic fluorocycline antibiotic in development.
Eravacycline (TP-434 or 7-fluoro-9-pyrrolidinoacetamido-6-demethyl-6-deoxytetracycline) is a novel fluorocycline that was evaluated for antimicrobial activity against panels of recent aerobic and anaerobic Gram-negative and Gram-positive bacteria. Eravacycline showed potent broad spectrum activity against 90% of the isolates (MIC90) in each panel at concentrations ranging from ≤0.008 to 2 μg/mL for all species panels except Pseudomonas aeruginosa and Burkholderia cenocepacia (MIC90 values of 32 μg/mL for both organisms). The antibacterial activity of eravacycline was minimally affected by expression of tetracycline-specific efflux and ribosomal protection mechanisms in clinical isolates. Further, eravacycline was active against multidrug-resistant bacteria, including those expressing extended spectrum β-lactamases and mechanisms conferring resistance to other classes of antibiotics, including carbapenem resistance. Eravacycline has the potential to be a promising new IV/oral antibiotic for the empiric treatment of complicated hospital/healthcare infections and moderate-to-severe community-acquired infections.
Tetraphase’s lead product candidate, eravacycline, has also received an award from Biomedical Advanced Research and Development Authority (BARDA) that provides for funding to develop eravacycline as a potential counter-measure to certain biothreat pathogens. It is worth up to USD 67 million.
Process R&D of Eravacycline: The First Fully Synthetic Fluorocycline in Clinical Development
Eravacycline (TP-434)
We are developing our lead product candidate, eravacycline, as a broad-spectrum intravenous and oral antibiotic for use as a first-line empiric monotherapy for the treatment of multi-drug resistant (MDR) infections, including MDR Gram-negative bacteria. We developed eravacycline using our proprietary chemistry technology. We completed a successful Phase 2 clinical trial of eravacycline with intravenous administration for the treatment of patients with complicated intra-abdominal infections (cIAI) and have initiated the Phase 3 clinical program.
Eravacycline is a novel, fully synthetic tetracycline antibiotic. We selected eravacycline for development from tetracycline derivatives that we generated using our proprietary chemistry technology on the basis of the following characteristics of the compound that we observed in in vitro studies of the compound:
- potent antibacterial activity against a broad spectrum of susceptible and multi-drug resistant bacteria, including Gram-negative, Gram-positive, atypical and anaerobic bacteria;
- potential to treat the majority of patients as a first-line empiric monotherapy with convenient dosing; and
- potential for intravenous-to-oral step-down therapy.
In in vitro studies, eravacycline has been highly active against emerging multi-drug resistant pathogens like Acinetobacter baumannii as well as clinically important species ofEnterobacteriaceae, including those isolates that produce ESBLs or are resistant to the carbapenem class of antibiotics, and anaerobes.
Based on in vitro studies we have completed, eravacycline shares a similar potency profile with carbapenems except that it more broadly covers Gram-positive pathogens like MRSA and enterococci, is active against carbapenem-resistant Gram-negative bacteria and unlike carbapenems like Primaxin and Merrem is not active against Pseudomanas aeruginosa. Eravacycline has demonstrated strong activity in vitro against Gram-positive pathogens, including both nosocomial and community-acquired methicillin susceptible or resistantStaphylococcus aureus strains, vancomycin susceptible or resistant Enterococcus faecium andEnterococcus faecalis, and penicillin susceptible or resistant strains of Streptococcus pneumoniae. In in vitro studies for cIAI, eravacycline consistently exhibited strong activity against enterococci and streptococci. One of the most frequently isolated anaerobic pathogens in cIAI, either as the sole pathogen or often in conjunction with another Gram-negative bacterium, is Bacteroides fragilis. In these studies eravacycline demonstrated activity against Bacteroides fragilis and a wide range of Gram-positive and Gram-negative anaerobes.
Key Differentiating Attributes of Eravacycline
The following key attributes of eravacycline, observed in clinical trials and preclinical studies of eravacycline, differentiate eravacycline from other antibiotics targeting multi-drug resistant infections, including multi-drug resistant Gram-negative infections. These attributes will make eravacycline a safe and effective treatment for cIAI, cUTI and other serious and life-threatening infections for which we may develop eravacycline, such as ABSSSI and acute bacterial pneumonias.
- Broad-spectrum activity against a wide variety of multi-drug resistant Gram-negative, Gram-positive and anaerobic bacteria. In our recently completed Phase 2 clinical trial of the intravenous formulation of eravacycline, eravacycline demonstrated a high cure rate against a wide variety of multi-drug resistant Gram-negative, Gram-positive and anaerobic bacteria. In addition, in in vitro studies eravacycline demonstrated potent antibacterial activity against Gram-negative bacteria, including E. coli; ESBL-producing Klebsiella pneumoniae; Acinetobacter baumannii; Gram-positive bacteria, including MSRA and vancomycin-resistant enterococcus, or VRE; and anaerobic pathogens. As a result of this broad-spectrum coverage, eravacycline has the potential to be used as a first-line empiric monotherapy for the treatment of cIAI, cUTI, ABSSSI, acute bacterial pneumonias and other serious and life-threatening infections.
- Favorable safety and tolerability profile. Eravacycline has been evaluated in more than 250 subjects in the Phase 1 and Phase 2 clinical trials that we have conducted. In these trials, eravacycline demonstrated a favorable safety and tolerability profile. In our recent Phase 2 clinical trial of eravacycline, no patients suffered any serious adverse events, and safety and tolerability were comparable to ertapenem, the control therapy in the trial. In addition, in the Phase 2 clinical trial, the rate at which gastrointestinal adverse events such as nausea and vomiting that occurred in the eravacycline arms was comparable to the rate of such events in the ertapenem arm of the trial.
- Convenient dosing regimen. In our recently completed Phase 2 clinical trial we dosed eravacycline once or twice a day as a monotherapy. Eravacycline will be able to be administered as a first-line empiric monotherapy with once- or twice-daily dosing, avoiding the need for complicated dosing regimens typical of multi-drug cocktails and the increased risk of negative drug-drug interactions inherent to multi-drug cocktails.
- Potential for convenient intravenous-to-oral step-down. In addition to the intravenous formulation of eravacycline, we are also developing an oral formulation of eravacycline. If successful, this oral formulation would enable patients who begin intravenous treatment with eravacycline in the hospital setting to transition to oral dosing of eravacycline either in hospital or upon patient discharge for convenient home-based care. The availability of both intravenous and oral administration and the oral step-down will reduce the length of a patient’s hospital stay and the overall cost of care.
Additionally, in February 2012, Tetraphase announced a contract award from the Biomedical Advanced Research and Development Authority (BARDA) worth up to $67 million for the development of eravacycline, from which Tetraphase may receive up to approximately $40 million in funding. The contract includes pre-clinical efficacy and toxicology studies; clinical studies; manufacturing activities; and associated regulatory activities to position the broad-spectrum antibiotic eravacycline as a potential empiric countermeasure for the treatment of inhalational disease caused by Bacillus anthracis, Francisella tularensis and Yersinia pestis.\
PAPER
Process research and development of the first fully synthetic broad spectrum 7-fluorotetracycline in clinical development is described. The process utilizes two key intermediates in a convergent approach. The key transformation is a Michael–Dieckmann reaction between a suitable substituted aromatic moiety and a key cyclohexenone derivative. Subsequent deprotection and acylation provide the desired active pharmaceutical ingredient in good overall yield.
Process R&D of Eravacycline: The First Fully Synthetic Fluorocycline in Clinical Development
FDA Grants QIDP Designation to Eravacycline, Tetraphase’s Lead Antibiotic Product Candidate
– Eravacycline designated as a QIDP for complicated intra-abdominal infection (cIAI) and complicated urinary tract infection (cUTI) indications –
July 15, 2013 08:30 AM Eastern Daylight Time
WATERTOWN, Mass.–(BUSINESS WIRE)–Tetraphase Pharmaceuticals, Inc. (NASDAQ: TTPH) today announced that the U.S. Food and Drug Administration (FDA) has designated the company’s lead antibiotic product candidate, eravacycline, as a Qualified Infectious Disease Product (QIDP). The QIDP designation, granted for complicated intra-abdominal infection (cIAI) and complicated urinary tract infection (cUTI) indications, will make eravacycline eligible to benefit from certain incentives for the development of new antibiotics provided under the Generating Antibiotic Incentives Now Act (GAIN Act). These incentives include priority review and eligibility for fast-track status. Further, if ultimately approved by the FDA, eravacycline is eligible for an additional five-year extension of Hatch-Waxman exclusivity.
/////////Tetraphase Pharmaceuticals, TP-434, 1207283-85-9, eravacycline
CN(C)C1C2CC3CC4=C(C=C(C(=C4C(=C3C(=O)C2(C(=C(C1=O)C(=O)N)O)O)O)O)NC(=O)CN5CCCC5)F