














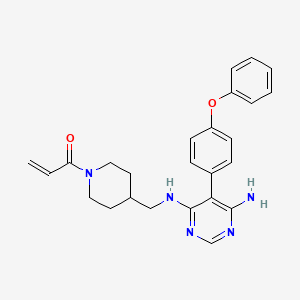
Evobrutinib
429.5 g/mol,C25H27N5O2
- Evobrutinib
- 1415823-73-2
- Evobrutinib [INN]
- 1-(4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl)piperidin-1-yl)prop-2-en-1-one
- MSC2364447C
- MSC2364447C
- M-2951
- MSC-2364447C
- ZA45457L1K
- 1-[4-[[[6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl]amino]methyl]piperidin-1-yl]prop-2-en-1-one
- M2951
Evobrutinib is under investigation in clinical trial NCT03934502 (Effect of Meal Composition and Timing on Evobrutinib Bioavailability).
Evobrutinib is an inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Upon administration, evobrutinib inhibits the activity of BTK and prevents the activation of the B-cell antigen receptor (BCR) signaling pathway. This prevents both B-cell activation and BTK-mediated activation of downstream survival pathways, which leads to the inhibition of the growth of malignant B-cells that overexpress BTK. BTK, a member of the Src-related BTK/Tec family of cytoplasmic tyrosine kinases, is overexpressed in B-cell malignancies; it plays an important role in B-lymphocyte development, activation, signaling, proliferation and survival.
Evobrutinib is in clinical development to investigate its potential as a treatment for multiple sclerosis (MS). It is an oral, highly selective inhibitor of Bruton’s tyrosine kinase (BTK) which is important in the development and functioning of various immune cells including B lymphocytes and macrophages.
Evobrutinib is designed to inhibit primary B cell responses such as proliferation and antibody and cytokine release, without directly affecting T cells. BTK inhibition is thought to suppress autoantibody-producing cells, which preclinical research suggests may be therapeutically useful in certain autoimmune diseases.
U.S. Patent No. 9073947 discloses a pyrimidine derivative of Evobrutinib which chemically named as l-(4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl)
piperidin-l-yl)prop-2-en-l-one and pharmaceutically acceptable salts, solvates and pharmaceutical compositions thereof.
U.S. Patent No. 9073947 and ‘Journal of Medicinal Chemistry 2019, 62(17), 7643-7655’ discloses process for the preparation of Evobrutinib which involves column purifications and lyophilisation methods to provide Evobrutinib with low yield, which is not viable at large scale production.
https://www.frontiersin.org/articles/10.3389/fnume.2021.820235/full
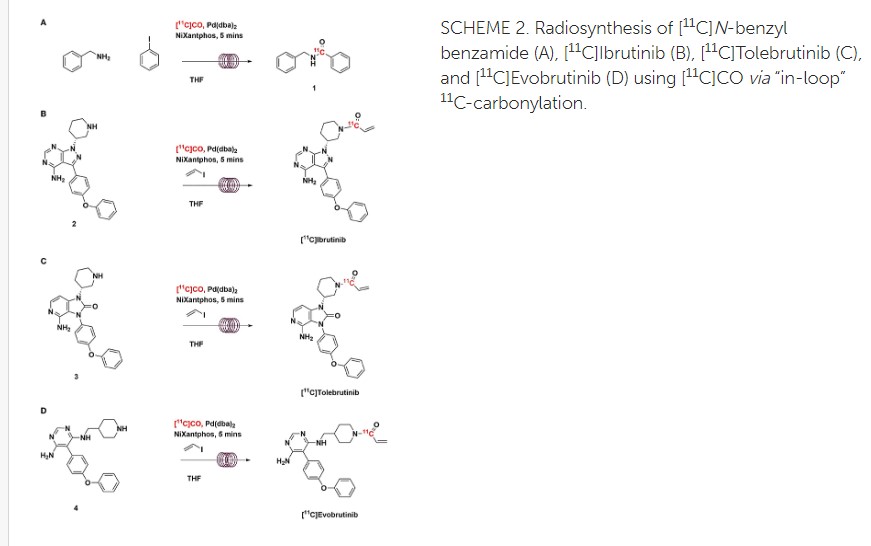
Radiosynthesis of [11C]Evobrutinib. [11C]Evobrutinib was synthesized similarly to the Tolebrutinib example above with the following exceptions. First, the precursor 5-(4-phenoxyphenyl)-N4-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine (4) (1 mg, 2.7 μmol) was used and the crude reaction mixture after the carbonylation reaction was purified by semi-preparative HPLC (column: Luna C18(2), 5 μ (250 x 9.6 mm); mobile phase: 44% MeCN in 200 mM ammonium formate; flow rate: 5 ml/min; UV: 254 nm). The [11C]1-(4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl)piperidin-1-yl)prop-2-en-1-one ([11C]evobrutinib) was isolated between the 15.5 and 18 min mark of the chromatogram and this sample was collected into a dilution flask that contained 50 ml of a 2 mg/ml sodium ascorbate aqueous solution. This solution was transferred to an HLB light (30 mg) SPE cartridge. After transfer, the cartridge was eluted with 1 ml of ethanol into the sterile product vial that contained 4 ml of sterile saline. Using this method, 2.2 ± 0.6 GBq (81.4 ± 22.2 mCi) [11C]evobrutinib was isolated (n = 3), and the product was analyzed via reverse phase HPLC using the following methods. Method A described above and Method B (Isocratic and molar activity): column: Luna C18(2) 3-μm (250×4.6 mm); mobile phase Isocratic: 36% acetonitrile in aqueous 0.1% TFA; flow rate: 1.3 ml/min; UV: 254 nm. Method A was used to confirm chemical identity using a co-injection of non-radioactive standard. Radiochemical purity and molar activity were determined by Method B. [11C]Evobrutinib was confirmed by co-injection with a verified non-radioactive reference standard. Am was determined using a 4-point standard curve (analytical HPLC peak area) (Y) vs. standard concentration (X: in nmol) by comparison with an evobrutinib reference standard of known concentration (2.3 mg in 1 ml). The isolated [11C] evobrutinib was co-eluted with a non-radioactive reference standard. The sample was >99% radiochemically pure, >95% chemically pure (HPLC, UV: 254 nm), with a molar activity of 496.5 ± 74 GBq/μmol (13.4 Ci/μmol) The overall synthesis time from the end of cyclotron bombardment was 37–46 min.
Patent
U.S. Patent No. 9073947
PAPER
Journal of Medicinal Chemistry 2019, 62(17), 7643-7655
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b00794
Step 4
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023157030&_gid=202334
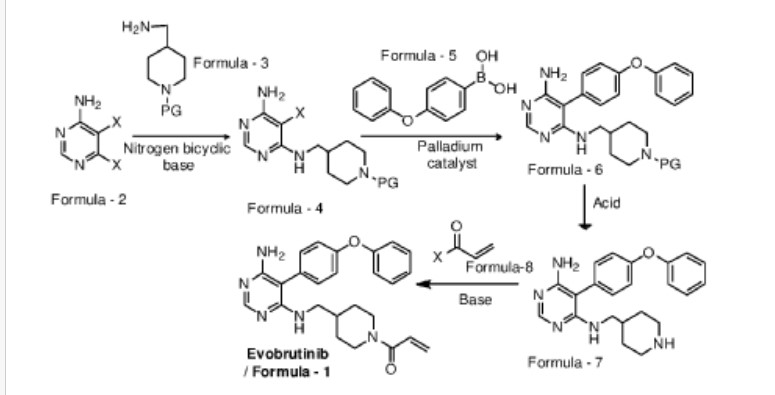
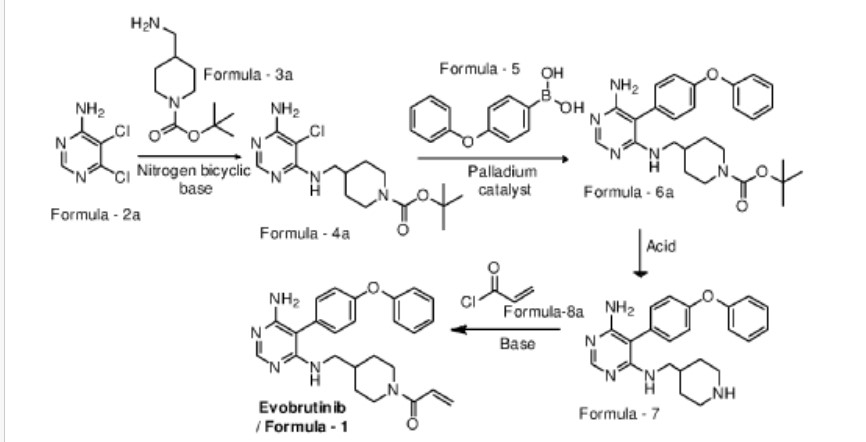
Examples:
Example-1: Preparation of tert-butyl 4-(((6-amino-5-chloropyrimidin-4-yl)amino) met hy 1 jpiperid ine- 1 -carboxylate
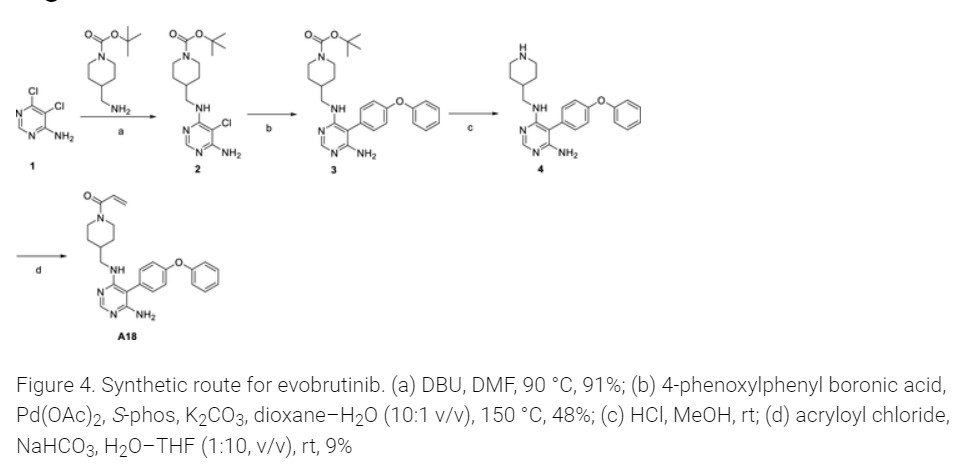
Tert-butyl-4-(aminomethyl)piperidine-l -carboxylate (81 ml) and 1,8-diazabicyclo [5.4.0]undec-7-ene (60.34 g) were added to a mixture of 5,6-dichloropyrimidin-4-amine (50 g) in N,N-dimethylformamide (500 ml) at 25-35°C. Heated the mixture to 90-95°C and stirred for 22 hrs. Cooled the mixture to 25-30°C. Water was added to the mixture at 25-35°C and stirred for 5 hrs. Filtered the precipitated solid, washed with water and n-heptane and dried to get the title compound. Yield: 73.0 gms; Purity by HPLC: 98.7%
Example-2: Preparation of tert-butyl 4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl) amino)methyl)piperidine-l-carboxylate
(4-Phenoxyphenyl)boronic acid (75.12 g) was added to a mixture of tert-butyl 4-(((6-amino-5-chloropyrimidin-4-yl)amino)methyl)piperidine-l-carboxylate(100 g), 2-di cyclo hexylphosphino-2′,6′-dimethoxybiphenyl (12 g) and potassium carbonate (121.28 g) in 1,4-di oxane (1000 ml) at 25-30°C and stirred for 30 minutes under nitrogen atmosphere. Palladium acetate (1.96 g) was added to the mixture at 25-30°C. Heated the mixture to 100-105°C and stirred for 3 hrs. Cooled the mixture to 25-30°C. Water and ethyl acetate were added to the mixture at 25-35°C and stirred for 30 minutes. Filtered the mixture by using hyflow bed. Organic layer was separated from the filtrate. Organic layer was treated with carbon powder and distilled-off the solvent under reduced pressure, n-heptane (800 ml) was added to the obtained compound. Heated the mixture to 60-65°C and stirred for 90 minutes. Cooled the mixture to 25-30°C and stirred for 2 hrs. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound. Yield: 120 gms, Purity by HPEC: 97.6% Example-3: Preparation of 5-(4-phenoxyphenyl)-N4-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine
Tert-butyl-4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl) piperidine- 1 -carboxylate (200 g) in methanol (600 ml) was cooled to 0-5°C. Hydrochloric acid in ethyl acetate (500 ml) was slowly added to the mixture at 0-5°C. Mixture allowed to warm to 25-30°C and stirred for 20 hours. Water was added to the mixture and treated the mixture with aqueous ammonia solution. Dichloromethane was added to the mixture at 25-30°C and stirred for 10 minutes. Layers were separated and distilled-off the organic layer under reduce pressure. Obtained compound was treated with isopropyl ether and dried to get the title compound. Yield: 150 gms, Purity by HPLC: 76.4%
Example-4: Preparation of Evobrutinib
Sodium bicarbonate (23.86 g) and water (301 ml) were added to the mixture of 5-(4-phenoxyphenyl)-N4-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine (70 g) in tetrahydrofuran (2800 ml). Cooled the mixture to 0-5°C. Acryloyl chloride (23.62 g) was slowly added to the mixture. Mixture allowed to warm to 25-30°C and stirred for 20 hrs. Distilled-off the solvent from the mixture under reduced pressure. Ethyl acetate and water were added to the mixture and stirred for 10 minutes. Both the layers were separated. Organic layer was treated with aqueous hydrochloric acid solution and carbon powder. Distilled-off the organic layer under reduced pressure. Isopropyl ether was added to the mixture at 25-30°C and stirred for 14 hrs. Filtered the mixture and washed with isopropyl ether. Dried to get the title compound.
Yield: 41.8 gms, Purity by HPLC: 97.6%



join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
//////////PHASE 3, MSC2364447C, M-2951, MSC-2364447C, ZA45457L1K, M2951, M 2951, Evobrutinib