DeveloperAbbisko Therapeutics; AstraZeneca; Dust Diseases Authority; Institute of Respiratory Health; National Cancer Institute (USA); University of Glasgow; University of Leeds; University of Wisconsin-Madison
ClassAntineoplastics; Benzamides; Phenyl ethers; Piperazines; Pyrazoles; Small molecules
Phase IIGastric cancer; Lymphoma; Multiple myeloma; Solid tumours; Urogenital cancer
PreclinicalSkin cancer
No development reportedLiver cancer
DiscontinuedBladder cancer; Breast cancer; Glioblastoma; Head and neck cancer; Lung cancer; Mesothelioma; Non-small cell lung cancer; Oesophageal cancer
13 Sep 2024Pharmacodynamics data from the preclinical studies in Solid tumours presented at the 49th European Society for Medical Oncology Congress (ESMO-2024)
28 Feb 2024No recent reports of development identified for preclinical development in Liver-cancer in China (PO)
23 Jan 2024Preclinical trials in Solid tumours (Monotherapy) in China (PO) (Abbisko Therapeutics pipeline, January 2024)
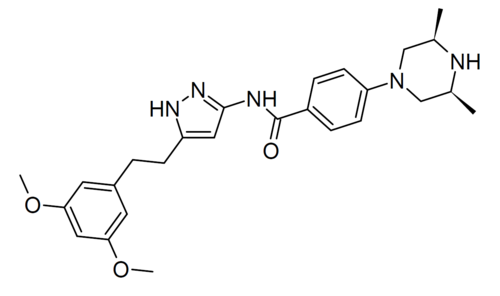
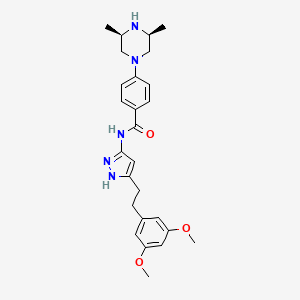
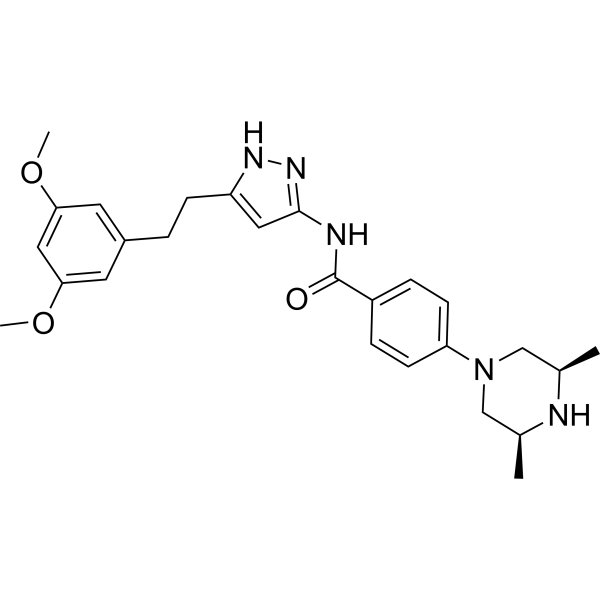
Fexagratinib (AZD4547) is an experimental drug which acts as an inhibitor of the fibroblast growth factorreceptors, having high affinity for FGFR1, FGFR2 and FGFR3 and weaker activity at FGFR4. It has reached clinical trials in humans against several forms of cancer, but has had only limited use as a medicine due to an unfavorable side effect profile, though it may have some applications in combination with other drugs. However it is still widely used in cancer research.[1][2][3][4][5]
SCHEME
SIDECHAIN
MAIN
SYN
At present, the preparation of AZD4547 mainly includes the following methods:
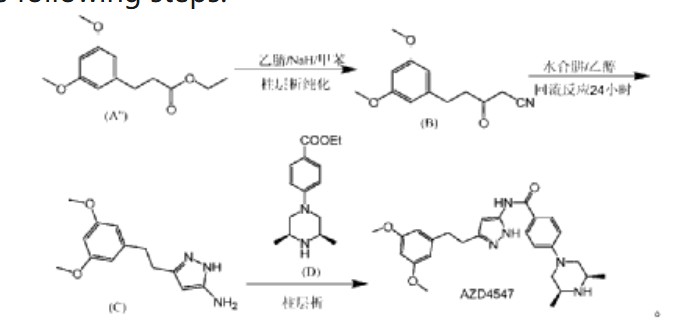
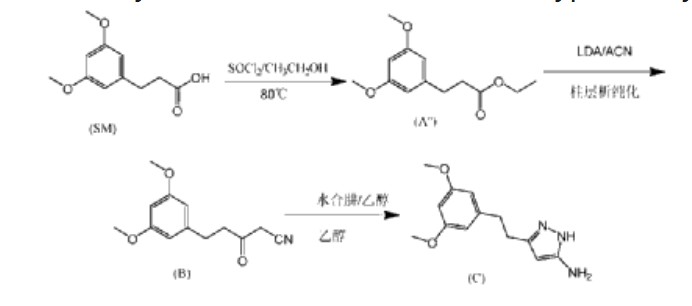
(1) Patent application WO2008075068A1 discloses a preparation method comprising the following steps:
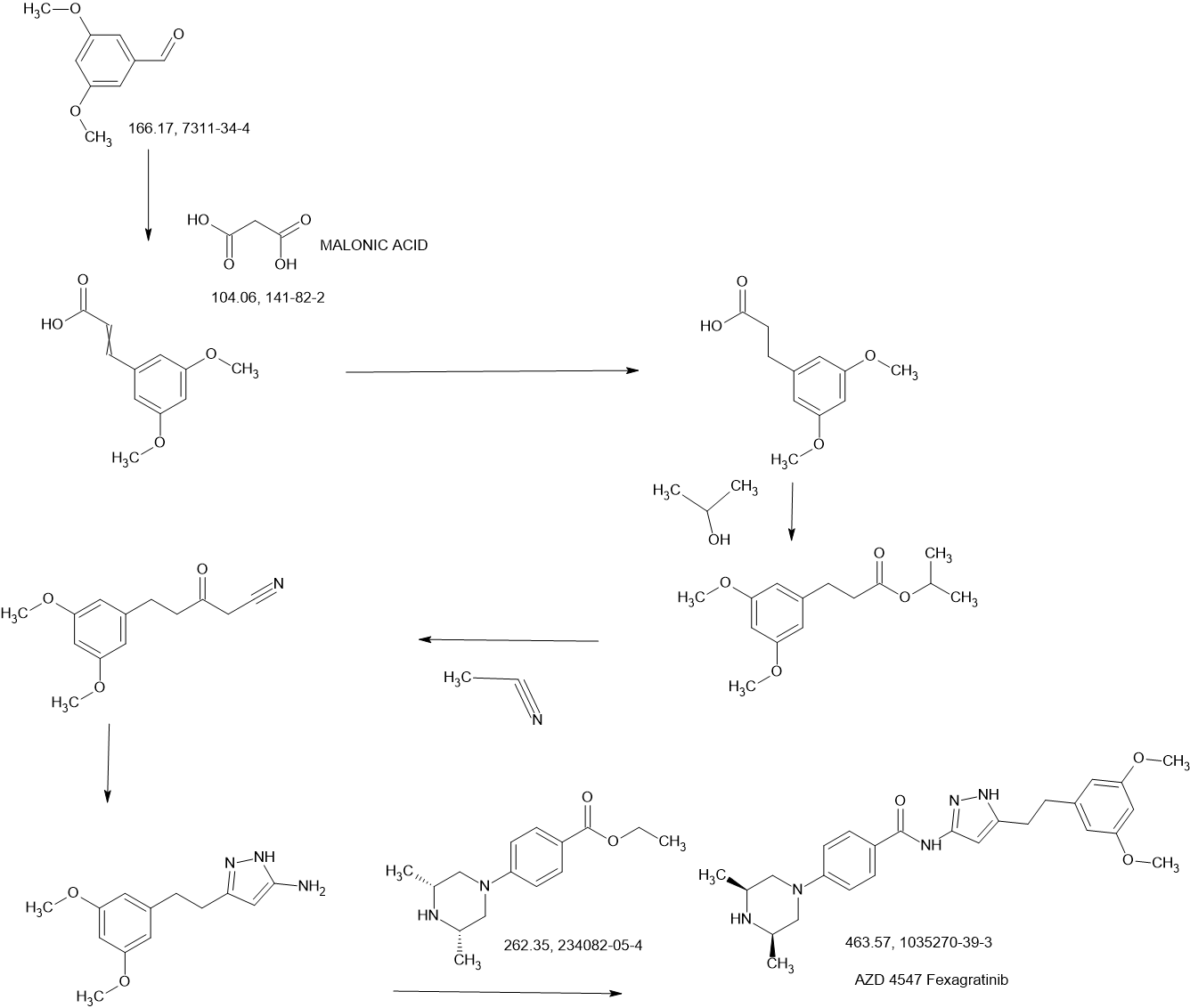
In the preparation method, AZD4547 is prepared by three-step reactions using ethyl 3-(3,5-dimethoxyphenyl)propionate as a raw material, wherein the first step reaction needs to be purified by column chromatography, and the yield is only 42%; the second step reaction requires reflux reaction for 24 hours, hydrazine hydrate is prone to explosion in high-temperature reactions, and hydrazine hydrate is a highly toxic and genotoxic reagent, and direct high-temperature reaction is not friendly to humans and the environment; the third step reaction also requires column chromatography purification, and the total yield of the three-step reaction for preparing AZD4547 is only 21.08%; therefore, the multi-step reactions of the preparation method require column chromatography operations, have poor safety, low yield, are not suitable for industrialization, and cannot solve the problem of drug accessibility.
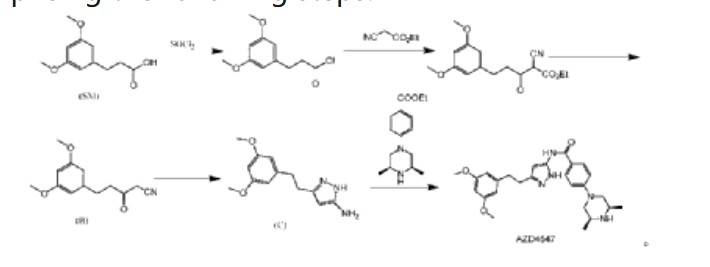
(2) Patent application CN111072638A discloses another preparation method, comprising the following steps:
In this preparation method, 3-(3,5-dimethoxyphenyl)propionic acid is used as the starting material, and AZD4547 is prepared through a five-step reaction with a total yield of 42.5%. In this preparation method, highly toxic reagent ethyl cyanoacetate and expensive reagents palladium carbon, stannous chloride, and Raney nickel are required, and it is not suitable for industrial production.
(3) In addition, patent application WO2016137506A1 discloses a method for preparing AZD4547 key intermediate 3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine, as follows:
In this preparation method, the first step of the reaction uses ethanol reflux reaction, the second step of the reaction uses a large amount of solvent, and needs to be reacted at an ultra-low temperature of -78°C. After the reaction is completed, column chromatography purification is required, which is not suitable for industrial application.
Add isopropanol (300 mL) and 3-(3,5-dimethoxyphenyl)propionic acid (60.0 g, 0.285 mol) to a 1L three-necked reaction bottle, raise the temperature to 40±5°C, and stir for 5 to 10 minutes to dissolve. Add SOCl dropwise at 40±5°C. 2 (37.3g, 0.314mol), the dropping time is ≥0.5 hours (the dropping process is obviously exothermic), after the dropping is completed, the temperature is raised to 60±5℃, the reaction is stirred for 1 hour, and the reaction of the raw materials is complete when HPLC is detected. The reaction solution is cooled to 35±5℃, the temperature is controlled below 50℃ and the solution is concentrated under reduced pressure until there is no obvious fraction, methyl tert-butyl ether (300mL) is added to dissolve, 5% potassium carbonate aqueous solution is added under ice bath to adjust the pH value of the reaction solution to 8-9, the temperature is controlled at 25±5℃ and stirred for 0.5 hours, the solution is allowed to stand and the organic phase is separated, washed with saturated brine, and the solution is concentrated under reduced pressure at 45℃ to dryness to obtain 72.1g of light yellow oily 3-(3,5-dimethoxyphenyl) propionic acid isopropyl ester, purity: 94%, yield: 94.3%.
Under nitrogen protection, add isopropyl 3-(3,5-dimethoxyphenyl)propionate (20.0 g, 0.079 mol), anhydrous acetonitrile (80 ml), and anhydrous tetrahydrofuran (100 ml) to a 500 ml three-necked reaction bottle, cool the reaction solution to an internal temperature of about -20 ° C, slowly add lithium diisopropylamide (83 ml, 0.166 mol, 2M THF solution), and add the solution dropwise for about 25 minutes. Continue stirring for 5-10 minutes, and detect the reaction of the raw materials by HPLC. After the reaction mixture was completely dried, acetic acid solution (15 ml) was added to quench the reaction, the mixture was concentrated under reduced pressure, water (100 ml) was added, the pH was adjusted to neutral with 25% aqueous sodium carbonate solution, ethyl acetate (200 ml) was added for extraction (HPLC chart see Figure 2), the organic layer was concentrated under reduced pressure until there was no fraction, ethanol (200 ml) was added, stirred and slurried, filtered, and the filter cake was dried in vacuum at 45°C to obtain 14.8 g of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile with a purity of 98% and a yield of 76%.
After preliminary separation, the HPLC, LCMS and 1 HNMR spectra of the impurity-containing mother liquor are shown in Figures 3-5. After analysis, the main impurity is generated by the self-polymerization of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile, and the structure of the impurity compound [compound of formula (B’)] is as follows:
Example 3
Under nitrogen protection, 3-(3,5-dimethoxyphenyl)propionic acid isopropyl ester (11.29 g, 0.045 mol), anhydrous acetonitrile (40 ml) and anhydrous tetrahydrofuran (50 ml) were added to a 500 ml three-necked reaction bottle, the reaction solution was cooled to -20°C, diisopropylamide lithium tetrahydrofuran solution (47 ml, 0.094 mol) was slowly added dropwise, and the addition was completed in about 25 minutes. The reaction was continued with stirring for 5-10 minutes. HPLC detected that the raw material reaction was complete, anhydrous ethanol (20 ml) was added to quench the reaction, and 2-methyltetrahydrofuran (50 g) was added for extraction. The pH of the aqueous layer was adjusted to neutral with hydrochloric acid, filtered, and the filter cake was dried in vacuo at 45°C to obtain 9.28 g of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile, purity: 99.5%, yield: 88.0%.
Example 4
Under nitrogen protection, a tetrahydrofuran solution containing isopropyl 3-(3,5-dimethoxyphenyl)propionate (120.0 g, 0.4756 mol), anhydrous acetonitrile (380 g, 9.25 mol), and anhydrous tetrahydrofuran (270 g) were added to a 3L three-necked reaction flask. The mixture was stirred until the internal temperature dropped to about -20°C. At this temperature, a tetrahydrofuran solution of lithium diisopropylamide (500 ml, 1 mol) was slowly added dropwise. After the addition was completed, the mixture was stirred at about -20°C for 1 minute. -2 hours, HPLC detected that the raw material was completely converted, ethanol (474g) was added to the reaction to quench the reaction, and the reaction was concentrated under reduced pressure. Purified water was added, the internal temperature was controlled at 0-15°C, HCl was slowly added, the pH was adjusted to 7.0, and a large amount of solid was precipitated. The reaction was stirred for 30 minutes and filtered, and the mixture was rinsed with purified water and ethanol in turn. The mixture was dried in vacuo at 45°C to obtain 98.7g of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile with a purity of 99.6% and a yield of 89.0%.
In addition, the inventors investigated the effects of the reaction raw materials, anhydrous acetonitrile, alkaline reagent, and reaction temperature on the reaction. The purity and reaction phenomena in the HPLC test were as follows:
From the above experimental investigation factors and experimental phenomena, it can be seen that the types of ester groups of different reaction raw materials, the amount of acetonitrile and the alkaline reagent have the following effects on the reaction:
(1) Effect of the type of ester group in the reaction raw materials on the reaction
When the reaction raw material is a compound of formula (A’) having a methyl ester group, a sticky mass will be formed during the reaction, affecting stirring, and the purity of the reaction is not high. Specifically, at the beginning of the reaction, a sticky mass appears in the reaction liquid, affecting stirring. As the reaction proceeds, the reaction liquid gradually becomes sticky, and even sticks to the wall, making it impossible to stir.
When the reaction raw material is a compound of formula (A”) having an ethyl ester group, the reaction purity is increased to 87%, but sticky lumps are still formed during the reaction, affecting stirring. The specific situation is similar to that when the reaction raw material is a compound of formula (A’) having a methyl ester group.
The appearance of viscous clumps during process scale-up can easily lead to incomplete reactions, and may even cause dangerous situations such as entanglement of stirring blades and burning of motors. Therefore, the above two preparation processes are not suitable for industrial scale-up production.
When the ester structure of the reaction raw material is changed to isopropyl ester, the reaction liquid is homogeneously clear without sticky micelles, and the reaction control purity is increased to more than 97%, which is suitable for industrial scale-up production. The inventors analyzed that the above experimental phenomenon may be due to the higher stability of the isopropyl ester structure, which reduces the formation of side reactions.
(2) Effect of acetonitrile dosage on the reaction
In Experiment 6 and Experiment 3 of the present invention, when the molar ratio of acetonitrile to the reaction raw material increased from 10eq to 20eq, the reaction control purity increased from 90.4% to 97.2%.
Comparing Experiment 2 and Experiment 3 in Experiment 1, when the molar ratio of acetonitrile to the reaction raw material increased from 1.2eq to 25eq, the reaction control purity increased from 60.8% to 92.8%.
(3) Effect of the selection and dosage of alkaline reagents on the reaction
From the above experimental results, it can be seen that the reaction control purity of NaHMDS, LDA and n-BuLi is relatively high.
The optimal molar ratio of alkaline reagent to reaction raw materials is 2.1eq. A molar ratio lower than 2eq may result in incomplete reaction.
Example 5
Step 1: Synthesis of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile (compound of formula (B))
Under nitrogen protection, add isopropyl 3-(3,5-dimethoxyphenyl)propionate (20.0 g, 0.079 mol), anhydrous acetonitrile (80 ml), and anhydrous tetrahydrofuran (100 ml) into a 500 ml three-necked reaction bottle, cool the reaction solution to -20°C, slowly add lithium diisopropylamide (83 ml, 0.166 mol, 2M THF solution) dropwise, add for about 25 min, stir and react for 5-10 minutes, HPLC detection shows that the raw material reaction is complete, add anhydrous ethanol (40 ml), concentrate under reduced pressure to a viscous state, add anhydrous ethanol (60 ml) to prepare an ethanol solution, and directly put into the next step reaction.
Step 2: Preparation of 3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine (compound of formula (C))
Add acetic acid (26.0 g, 0.436 mol), ethanol (100 ml), and 80% hydrazine hydrate (15.0 g, 0.238 mol) to a 500 ml three-necked reaction bottle, heat to an internal temperature of about 68°C, slowly add the product ethanol solution (18.5 g, 0.079 mol) obtained in step 1 to the mixed solution of acetic acid and hydrazine hydrate at this temperature, add for about 40 minutes, stir and react at an internal temperature of about 68°C for 1 hour, and HPLC detection shows that 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile is completely converted; the reaction solution is concentrated under reduced pressure, water (100 ml), ethyl acetate (200 ml), and about 25% Na 2 CO 3 (40ml) adjust the pH value of the water layer to 7-8; separate the water layer, wash the layers with saturated brine (20ml), concentrate the organic layer under reduced pressure until there is no fraction, add isopropyl acetate (100ml) and reduce the pressure to bring it to a viscous state, add isopropyl acetate (120ml) and heat to dissolve, cool and crystallize, filter at about 10°C, and dry under vacuum at 50°C to obtain 16.3g of 3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine with a purity of 99.6% and a total yield of 83% in two steps.
In addition, the inventors investigated the effect of the amount of acetic acid used in this step of the reaction on the reaction, and the purity was controlled by HPLC as follows:
In addition, the inventors also investigated that the solid compound of formula (B) obtained after purification of the product in step 1 was reacted with hydrazine hydrate in the presence of acetic acid, and the compound of formula (B) was also completely converted to obtain a high-purity compound of formula (C).
Example 6
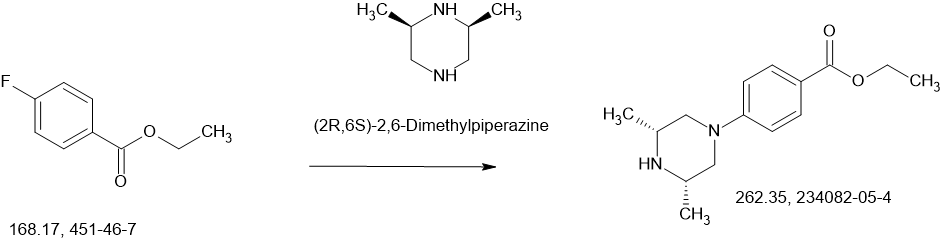
3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine (100.0 g, 0.4044 mol), ethyl 4-((3R,5S)-3,5-dimethylpiperazin-1-yl)benzoate (132.5 g, 0.5050 mol), and 2-methyltetrahydrofuran (1300 ml) were added to the reaction kettle, heated to 50-55°C and stirred for 1 hour, filtered through diatomaceous earth, and the filtrate was added to a clean reaction kettle, heated to atmospheric distillation with water, and the reaction temperature was controlled at 78°C-88°C, and 25% KO-tAm toluene solution (490.0 g) was slowly added dropwise for about 2 hours. After the addition was completed, the reaction temperature was adjusted to 83-88°C and stirred for 3-6 hours. Sampling was performed to detect whether the reaction of the raw materials was complete. The reaction system was cooled to 30-60°C, water (8 ml) was slowly added to quench the reaction, and the mixture was stirred at 30-60°C for 0.5 hour, then cooled to about 25°C, water (400 ml) was added, stirred and allowed to stand for stratification, the organic phase was separated, water (200 ml) was added, the mixture was heated to about 50°C and stirred for 0.5 hour, the water layer was separated, and this was repeated 2-3 times until the pH of the water layer was 7.0-9.5; the organic layer was concentrated under reduced pressure to remove part of the solvent, the residue was heated to 80-90°C and stirred for 1 hour, slowly cooled to 20-30°C, stirred for 2-5 hours, filtered, rinsed twice with ethyl acetate, and dried in vacuo at 45°C to obtain 155.6 g of a white amorphous solid product (AZD4547) with a purity of 98.5% and a yield of 83%.
3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine (10.0 g, 0.040 mol), ethyl 4-((3R,5S)-3,5-dimethylpiperazin-1-yl)benzoate (12.1 g, 0.047 mol), anhydrous tetrahydrofuran (170 ml) were added to the reaction bottle, heated and distilled at atmospheric pressure until about 100 ml remained, cooled to -30°C to -20°C, and NaHMDS (0.125 mol, 63 ml, 2M THF solution) was slowly added dropwise. The temperature of the reaction system was controlled at about -25°C and stirred for 20 minutes. HPLC detected that the raw materials were basically reacted. Water (30 ml) was slowly added under temperature control to quench, and glacial acetic acid (about 10 ml) was added to neutralize. The temperature was raised to about 0°C and stirred, and 20% Na 2 CO 3 (10ml), concentrated under reduced pressure until there is no fraction, added ethyl acetate (120ml) to the residue, heated to about 45°C, stirred and separated, the organic phase was separated, added with saturated brine (30ml), washed once, concentrated under reduced pressure to leave about (30ml), then added ethyl acetate (30ml), concentrated under reduced pressure again, repeated twice, a large amount of solid precipitated, added ethyl acetate to a material volume of about 50ml, stirred at 0-10°C for 1 hour, filtered, and dried in vacuo at 45°C to obtain 16.87g of white amorphous solid product (AZD4547) with a purity of 99.8% and a yield of 91%.
Heterocycles (2020), 100(2), 276-282 ,
CN111072638
PATENT
CN115819239
Nature Catalysis (2021), 4(5), 385-394
Shandong Huagong (2021), 50(7), 19-21
CN111072638
Heterocycles (2020), 100(2), 276-282
Physical Chemistry Chemical Physics (2020), 22(17), 9656-9663
Journal of Chemical Theory and Computation (2019), 15(2), 1265-1277
Journal of Medicinal Chemistry (2017), 60(14), 6018-6035
^ Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, et al. (April 2012). “AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family”. Cancer Research. 72 (8): 2045–2056. doi:10.1158/0008-5472.CAN-11-3034. PMID22369928.
^ Katoh M, Nakagama H (March 2014). “FGF receptors: cancer biology and therapeutics”. Medicinal Research Reviews. 34 (2): 280–300. doi:10.1002/med.21288. PMID23696246.
^ Zengin ZB, Chehrazi-Raffle A, Salgia NJ, Muddasani R, Ali S, Meza L, et al. (February 2022). “Targeted therapies: Expanding the role of FGFR3 inhibition in urothelial carcinoma”. Urologic Oncology. 40 (2): 25–36. doi:10.1016/j.urolonc.2021.10.003. PMID34840077.















{kind=link}