















54143-56-5
54143-55-4 (free base)
- Molecular FormulaC17H20F6N2O3
- Average mass414.343 Da

R-818
Flecainide is a medication used to prevent and treat abnormally fast heart rates.[1] This includes ventricular and supraventricular tachycardias.[1] Its use is only recommended in those with dangerous arrhythmias or when significant symptoms cannot be managed with other treatments.[1] Its use does not decrease a person’s risk of death.[1] It is taken by mouth or injection into a vein.[1][2]
Common side effects include dizziness, problems seeing, shortness of breath, chest pain, and tiredness.[1] Serious side effects may include cardiac arrest, arrhythmias, and heart failure.[1] It may be used in pregnancy, but has not been well studied in this population.[2][3] Use is not recommended in those with structural heart disease or ischemic heart disease.[1] Flecainide is a class Ic antiarrhythmic agent.[1] It works by decreasing the entry of sodium in heart cells, causing prolongation of the cardiac action potential.[1]
Flecainide was approved for medical use in the United States in 1985.[1] It is available as a generic medication.[2] A month supply in the United Kingdom costs the NHS about £7.68 as of 2019.[2] In the United States the wholesale cost of this amount is about 18.60 USD.[4]In 2016 it was the 273rd most prescribed medication in the United States with more than a million prescriptions.[5]
Medical uses
Flecainide is used in the treatment of many types of supraventricular tachycardias, including AV nodal re-entrant tachycardia (AVNRT) and Wolff-Parkinson-White syndrome (WPW).
It also has limited use in the treatment of certain forms of ventricular tachycardia (VT). In particular, flecainide has been useful in the treatment of ventricular tachycardias that are not in the setting of an acute ischemic event. It has use in the treatment of right ventricular outflow tract (RVOT) tachycardia[6] and in the suppression of arrhythmias in arrhythmogenic right ventricular dysplasia (ARVD).[7]Studies (notably the Cardiac Arrhythmia Suppression Trial) have shown an increased mortality when flecainide is used to suppress ventricular extrasystoles in the setting of acute myocardial infarction.[8][9]
In individuals suspected of having the Brugada syndrome, the administration of flecainide may help reveal the ECG findings that are characteristic of the disease process. This may help make the diagnosis of the disease in equivocal cases.[10]
Flecainide has been introduced into the treatment of arrhythmias in children.
In the long-term, flecainide seems to be safe in people with a healthy heart with no signs of left ventricular hypertrophy, ischemic heart disease, or heart failure.[11]
Side effects
Results of a medical study known as the Cardiac Arrhythmia Suppression Trial (CAST) demonstrated that patients with structural heart disease (such as a history of MI (heart attack), or left ventricular dysfunction) and also patients with ventricular arrhythmias, should not take this drug. The results were so significant that the trial was stopped early and preliminary results were published.[12]
The dose may need to be adjusted in certain clinical scenarios. As with all other antiarrhythmic agents, there is a risk of proarrhythmiaassociated with the use of flecainide. This risk is probably increased when flecainide is co-administered with other class Ic antiarrhythmics, such as encainide. The risk of proarrhythmia may also be increased by hypokalemia.[13] The risk of proarrhythmia is not necessarily associated with the length of time an individual is taking flecainide, and cases of late proarrhythmia have been reported.[14] Because of the role of both the liver and the kidneys in the elimination of flecainide, the dosing of flecainide may need to be adjusted in individuals who develop either liver failure or renal failure.
Because of the negative inotropic effects of flecainide, it should be used with caution in individuals with depressed ejection fraction, and may worsen congestive heart failure in these individuals. It should be avoided in people with ischaemic heart disease and the elderly.[15]
As with all class I antiarrhythmic agents, Flecainide increases the capture thresholds of pacemakers.[16]
Heart
Due to the narrow therapeutic index of flecainide, physicians should be alert for signs of toxicity before life-threatening arrhythmias occur like torsades de pointes. While the toxic effects of flecainide are closely related to the plasma levels of the drug,[17] it is unfeasible to check the plasma concentration in an individual on a regular basis.
Signs of flecainide toxicity include marked prolongation of the PR interval and widening of the QRS duration on the surface ECG. There may be signs and symptoms attributable to overt heart failure secondary to sudden decreased myocardial contractility.
Treatment
Treatment of flecainide cardiac toxicity involves increasing the excretion of flecainide, blocking its effects in the heart, and (rarely) institution of cardiovascular support to avoid impending lethal arrhythmias. Modalities that have had success include administration of a beta-sympathomimetic agent,[17] and administration of a sodium load[17](often in the form of hypertonic sodium bicarbonate). Placing the individual on cardiopulmonary bypass support may be necessary in order to temporarily remove the need for a beating heart and to increase blood flow to the liver.[18][19]
Lungs
Flecainide has a very high affinity for lung tissue [20] and is associated with drug-induced interstitial lung disease.[21][22][23][24][25]
Interactions
Flecainide has high bioavailability after an oral dose,[26] meaning that most of the drug that is ingested will enter the systemic blood stream. Peak serum concentrations can be seen 1 to 6 hours after ingestion of an oral dose. While the plasma half-life is about 20 hours, it is quite variable, and can range from 12 to 27 hours.[27] During oral loading with flecainide, a steady state equilibrium is typically achieved in 3 to 5 days.
The majority of flecainide is eliminated by the kidneys, with the remainder metabolized by the cytochrome P450 2D6 isoenzyme in the liver.[28] Therefore, alterations in renal function or urine pH will greatly affect the elimination of flecainide, as more is eliminated by the kidney than by the hepatic route.
Because of the dual elimination routes of flecainide and its tendency to decrease myocardial contractility,[15] flecainide interacts with numerous pharmaceuticals and can potentiate the effects of other myocardial depressants and AV node blocking agents. In addition, flecainide can decrease the metabolism or elimination of many (but not all) agents that use the cytochrome P450 enzyme system.
A full list of drug interactions with flecainide can be obtained from the manufacturer. Some important drug interactions with flecainide include:[citation needed]
- Alcohol – may further depress normal heart function
- Amiodarone – inhibits cytochrome P450 2D6 and may increase flecainide levels
- Cimetidine – increases flecainide levels by 30% and half-life by 10%
- Digoxin – may increase digoxin levels
- Paroxetine – increased effect of both drugs
- Propafenone – increased effect of both drugs and increased risk of toxicity
- Quinidine – inhibits cytochrome P450 2D6 and may increase flecainide levels
Overdose
Flecainide intoxication is rare but serious due to the cardiogenic shock that it provokes. Its diagnosis can be difficult in the lack of contributing anamnestic elements. Clinical and paraclinical signs are not specific. Treatment is primarily symptomatic, which gives good results thanks to the hypertonic solution of sodium salts. Organ donation is possible in the case of braindead patients who suffered a flecainide intoxication.[29]
Mechanism of action
Flecainide works by blocking the Nav1.5 sodium channel in the heart, slowing the upstroke of the cardiac action potential.[30] This thereby slows conduction of the electrical impulse within the heart, i.e. it “reduces excitability”. The greatest effect is on the His-Purkinje system and ventricular myocardium. The effect of flecainide on the ventricular myocardium causes decreased contractility of the muscle, which leads to a decrease in the ejection fraction.
The effect of flecainide on the sodium channels of the heart increases as the heart rate increases; This is known as use-dependence and is why that flecainide is useful to break a tachyarrhythmia.[31]
Flecainide also inhibits ryanodine receptor 2 (RyR2),[32] a major regulator of sarcoplasmic release of stored calcium ions. It can reduce calcium sparks and thus arrhythmogenic calcium waves in the heart.[33] While Flecainide therapy has been shown to suppress ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia(CPVT) and mouse models of this disease, the relative contribution from the inhibition of sodium channels and of RyR2 in this effect on CPVT is unclear.[34]
Brand names
Flecainide is sold under the trade name Tambocor (manufactured by 3M pharmaceuticals). Flecainide went off-patent on February 10, 2004. In addition to being marketed as Tambocor, it is also available in generic version and under the trade names Almarytm, Apocard, Ecrinal, and Flécaine.
Following is one of the synthesis routes: 2-Aminomethylpyridine (II) is condensed with 2,2,2-trifluoroethyl-2,5-bis(2,2,2-trifluoroethoxy)benzoate (I) in refluxing glyme to produce 2,5-bis(2,2,2-trifluoroethoxy)-N-(2-pyridylmethyl)benzamide (III), and the yielding product is then hydrogenated with H2 over Pd/C in acetic acid.

PATENT
https://patents.google.com/patent/US7196197B2/en
Flecainide acetate, 2,5-bis(2,2,2-trifluoroethoxy)-N-(2-piperidylmethyl)benzamide acetate (I), is a drug for the treatment of arrhythmia. It and its neutral base are described in U.S. Pat. No. 3,900,481.

A key intermediate for the synthesis of Flecainide and its pharmaceutically acceptable salts is 2,5-bis(2,2,2-trifluoroethoxy)benzoic acid (II). One prior method for the preparation of this intermediate, disclosed in British patent No. GB 2045760, is a multistep process which comprises the preparation of 1,4-bis(2,2,2-trifluoroethoxy)benzene from hydroquinone using the very expensive reagent trifluoroethyltriflate (CF3CH2OSO2CF3). 1,4-bis(2,2,2-trifluoroethoxy)benzene is then converted to 2,5-bis(2,2,2-trifluoroethoxy)benzoic acid (II) through a multistep process. An alternative method described in the same patent begins from 1,4-dibromobenzene, which is then condensed with more than 8 equivalents of 2,2,2-trifluoroethanol, to furnish the 1,4-bis(2,2,2-trifluoroethoxy)benzene intermediate. 2,5-bis(2,2,2-trifluoroethoxy)benzoic acid (II) is also be prepared starting from 1-bromo-4-fluorobenzene (PCT WO 02/066413) or from 2-bromo-5-chlorobenzoic acid (PCT WO 99/02498). All these approaches have limited commercial utility due to the cost of the reagents and the necessity for specialized equipment.
The method disclosed in British patent No. GB 2045760 for the preparation of the Flecainide base starts from 2,5-bis(2,2,2-trifluoroethoxy)benzoic acid which is converted to its acid chloride and reacts either with 2-(aminomethyl)piperidine to form Flecainide in one step or with 2-(aminomethyl)pyridine, followed by catalytic hydrogenation of the pyridine ring, to form Flecainide base in two steps. The disadvantage of the one step process is that the acid chloride reacts non-selectively with both nitrogen atoms of the 2-(aminomethyl)piperidine, resulting in a mixture of the two acylated isomers.
Other preparations of Flecainide base are disclosed in WO 99/02498 and US2003/0032835. The process disclosed in WO 99/02498 starts from the cyanomethyl ester of 2,5-bis(2,2,2-trifluoroethoxy)benzoic acid, which selectively reacts with the primary amino group of 2-(aminomethyl)piperidine to furnish Flecainide. US 2003/032835 discloses a procedure which involves converting 2,5-bis(2,2,2-trifluoroethoxy)benzoic acid to its activated 2,2,2-trifluoroethyl ester which then selectively reacts with the primary amino group of 2-(aminomethyl)piperidine to furnish Flecainide. Although activated esters of this type can be used for the formation of Flecainide, the reagents required to prepare them are expensive on the industrial scale. Moreover, the resulting cyanomethanol and 2,2,2-trifluoroethanol by-products are highly toxic. Esters from less expensive, non-toxic and readily available alcohols are still desired for commercial purposes. Based on the above deficiencies, a new process overcoming these deficiencies was required.





EXAMPLE 1
Preparation of 2-(2,2,2-trifluoroethoxy)benzoic acid
To a solution of 2,2,2-trifluoroethanol (40.0 g) and DMF (100 ml) was added sodium tert-butoxide (23.0 g) at 0° C. The solution was stirred at 20 to 25° C. for 1 hour at which point 2-chlorobenzoic acid (25.0 g) was added followed by cupric bromide (2.0 g). The mixture was stirred at 120° C. for 5 hours, cooled to 10° C., and water (30 ml) was added followed by 20% HCl solution (90 ml). The solution was extracted with dichloromethane (3×50 ml). The combined organic layers were washed with water (3×50 ml) and the volume was concentrated to 90 ml. Hexane (150 ml) was added to the residues, and the mixture was concentrated to volume of 120 ml and a further portion of hexane (30 ml) was added. The mixture was heated at 50° C. for 30 minutes and then stirred at room temperature for 1 hour. The solids were filtered to yield the crude product. This material was dissolved in ethyl acetate (50 ml), charcoal (1.7 g) was added and the mixture was stirred at room temperature a further 2 hours. The solution was filtered through Celite™ and crystallized from ethyl acetate/hexane to yield the pure product (30.9 g, yield 88.0%) as a white solid, m.p. 85–86° C.
EXAMPLE 2
Preparation of 5-bromo-2-(2,2,2-trifluoroethoxy)benzoic acid
To a solution of 2-(2,2,2-trifluoroethoxy)benzoic acid (22 g) in methylene chloride (100 ml), was added AlCl3 (13.3 g) at 0° C.followed by bromine (16.0 g, 0.1 mol). The reaction mixture was stirred at 0° C. for 1 hour and then at reflux for 2 hours. The solids were filtered and water (50 ml) and ethyl acetate (50 ml) were added to the filtrate. The aqueous layer was separated and extracted with ethyl acetate (2×60 ml) and the combined organic layers were washed with water (2×60 ml). The organic layer was concentrated under vacuum to dryness and hexane (100 ml) was added and the resulting suspension was stirred at 20 to 25° C. for 1 hour. The mixture was filtered and the cake was rinsed with heptanes (2×20 ml). The damp solids were dried in vacuum at 45° C. for 5–6 hours to give a white solid (28.3 g, yield 94.6%), m.p. 126–128° C.
EXAMPLE 3
Preparation of 2,5-bis(2,2,2-trifluoroethoxy)benzoic acid.
To a solution of 2,2,2-trifluoroethanol (14.7 g) and DMF (125 ml) was added sodium tert-butoxide (12.8 g) at 0° C. The solution was stirred at 20 to 25° C. for 1 hour at which point 5-bromo-2-(2,2,2-trifluoroethoxy)benzoic acid (20 g) was added followed by cupric bromide (2.0 g). The mixture was stirred at 100° C. for 10 hours, cooled to 10° C., and water (30 ml) was added followed by 20% HCl solution (90 ml). The solution was extracted with dichloromethane (3×80 ml), and the combined organic layers were washed with water (3×60 ml). The solution was concentrated to one-third of the original volume and hexane (200 ml) was added. The resulting suspension was stirred at room temperature for 2 hours, filtered and the damp cake was rinsed with hexane (2×40 ml). The damp cake was dried in vacuo at 40° C. for 5 hours to give the product as a white solid (16.02 g, yield 75.3%).
EXAMPLE 4
Preparation of methyl 2,5-bis(2,2,2-trifluoroethoxy)benzoate
A solution of 2,5-bis(2,2,2-trifluoroethoxy)benzoic acid (20 g) and thionyl chloride (15.0 g) in methanol (100 ml) was stirred at 80° C. for 2 hours. The solvents were evaporated under vacuum to give an oil residue. Toluene (100 ml) was added to the residue and the solution was washed with saturated NaHCO3 (30 ml) solution followed by water (3×30 ml). The organic layer was concentrated under reduced pressure to give the product as a white solid (20.5 g, yield 98.0%).
EXAMPLE 5
Preparation of Flecainide
A mixture of methyl 2,5-bis(2,2,2-trifluoroethoxy)benzoate (1.5 g), 2-(aminomethyl)piperidine (0.62 g) in toluene (3 ml) was stirred at reflux for 10 hours. After cooling to room temperature, water (10 ml) was added and two layers solution were separated. The aqueous layer was extracted with toluene (2×10 ml) and the combined organic layers were washed with water (3×10 ml). The organic layer was concentrated under reduced pressure to give Flecainide free base as a white solid (1.63 g, 85%).
EXAMPLE 6
Preparation of Flecainide acetate
To a solution of Flecainide free base (1.5 g) in isopropanol (7.5 ml) was added glacial acetic acid (0.3 g) and the solution was stirred under reflux for 2 hours. The solution was cooled to room temperature and hexane (15 ml) was added and solids began to precipitate. The resulting suspension was stirred at 20–25° C. for 2 hours and the solids were filtered and then rinsed with hexane (2×10 ml). The damp cake was dried in vacuum for 4 hours to give Flecainide acetate as a white solid (1.54 g, Yield 89%).
Patent
Flecainide
- ATC:C01BC04
- MW:414.35 g/mol
- CAS-RN:54143-55-4
- InChI Key:DJBNUMBKLMJRSA-UHFFFAOYSA-N
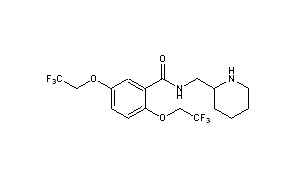
- InChI:InChI=1S/C17H20F6N2O3/c18-16(19,20)9-27-12-4-5-14(28-10-17(21,22)23)13(7-12)15(26)25-8-11-3-1-2-6-24-11/h4-5,7,11,24H,1-3,6,8-10H2,(H,25,26)
Derivatives
acetate
- Formula:C17H20F6N2O3 • C2H4O2
- MW:474.40 g/mol
- CAS-RN:54143-56-5
Synthesis Path
References
-
- Banitt, E.H. et al.: J. Med. Chem. (JMCMAR) 18, 1130 (1975); 20, 821 (1977).
- DE 2 513 916 (Riker; prior. 27.3.1975).
- US 3 900 481 (Riker; 19.8.1975; prior. 1.4.1974).
- US 4 005 209 (Riker; 25.1.1977; USA-prior. 1.4.1974, 27.5.1975).
References
- ^ Jump up to:a b c d e f g h i j k “Flecainide Acetate Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 7 April 2019.
- ^ Jump up to:a b c d British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. p. 103. ISBN 9780857113382.
- ^ “Flecainide (Tambocor) Use During Pregnancy”. Drugs.com. Retrieved 7 April 2019.
- ^ “NADAC as of 2019-02-27”. Centers for Medicare and Medicaid Services. Retrieved 3 March 2019.
- ^ “The Top 300 of 2019”. clincalc.com. Retrieved 22 December 2018.
- ^ Gill J, Mehta D, Ward D, Camm A (1992). “Efficacy of flecainide, sotalol, and verapamil in the treatment of right ventricular tachycardia in patients without overt cardiac abnormality”. Br Heart J. 68 (4): 392–97. doi:10.1136/hrt.68.10.392. PMC 1025139. PMID 1449923.
- ^ Sakurada H, Hiyoshi Y, Tejima T, Yanase O, Tokuyasu Y, Watanabe K, Motomiya T, Sugiura M, Hiraoka M (1990). “[Effects of oral flecainide treatment of refractory tachyarrhythmias]”. Kokyu to Junkan. 38 (5): 471–76. PMID 2115193.
- ^ Echt D, Liebson P, Mitchell L, Peters R, Obias-Manno D, Barker A, Arensberg D, Baker A, Friedman L, Greene H (1991). “Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial”. N Engl J Med. 324 (12): 781–88. doi:10.1056/NEJM199103213241201. PMID 1900101.
- ^ Greenberg H, Dwyer E, Hochman J, Steinberg J, Echt D, Peters R (1995). “Interaction of ischaemia and encainide/flecainide treatment: a proposed mechanism for the increased mortality in CAST I”. Br Heart J. 74 (6): 631–35. doi:10.1136/hrt.74.6.631. PMC 484119. PMID 8541168.
- ^ Gasparini M, Priori S, Mantica M, Napolitano C, Galimberti P, Ceriotti C, Simonini S (2003). “Flecainide test in Brugada syndrome: a reproducible but risky tool”. Pacing Clin Electrophysiol. 26 (1 Pt 2): 338–41. doi:10.1046/j.1460-9592.2003.00045.x. PMID 12687841.
- ^ Aliot E, Capucci A, Crijns HJ, Goette A, Tamargo J (2011). “Twenty-five years in the making: flecainide is safe and effective for the management of atrial fibrillation”. Eurospace. 13 (2): 161–73. doi:10.1093/europace/euq382. PMC 3024037. PMID 21138930.
- ^ Cardiac Arrhythmia Suppression Trial (CAST) Investigators (1989). “Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction”. N Engl J Med. 321 (6): 406–12. doi:10.1056/NEJM198908103210629. PMID 2473403.
- ^ Ohki R, Takahashi M, Mizuno O, Fujikawa H, Mitsuhashi T, Katsuki T, Ikeda U, Shimada K (2001). “Torsades de pointes ventricular tachycardia induced by mosapride and flecainide in the presence of hypokalemia”. Pacing Clin Electrophysiol. 24 (1): 119–21. doi:10.1046/j.1460-9592.2001.00119.x. PMID 11227957.
- ^ Morganroth J (1992). “Early and late proarrhythmia from antiarrhythmic drug therapy”. Cardiovasc Drugs Ther. 6 (1): 11–14. doi:10.1007/BF00050910. PMID 1533532.
- ^ Jump up to:a b Santinelli V, Arnese M, Oppo I, Matarazzi C, Maione S, Palma M, Giunta A (1993). “Effects of flecainide and propafenone on systolic performance in subjects with normal cardiac function”. Chest. 103 (4): 1068–73. doi:10.1378/chest.103.4.1068. PMID 8131440.
- ^ Fornieles-Pérez H, Montoya-García M, Levine P, Sanz O (2002). “Documentation of acute rise in ventricular capture thresholds associated with flecainide acetate”. Pacing Clin Electrophysiol. 25 (5): 871–72. doi:10.1046/j.1460-9592.2002.00871.x. PMID 12049386.
- ^ Jump up to:a b c Winkelmann B, Leinberger H (1987). “Life-threatening flecainide toxicity. A pharmacodynamic approach”. Annals of Internal Medicine. 106 (6): 807–14. doi:10.7326/0003-4819-106-6-807. PMID 3107447.
- ^ Corkeron M, van Heerden P, Newman S, Dusci L (1999). “Extracorporeal circulatory support in near-fatal flecainide overdose”. Anaesth Intensive Care. 27 (4): 405–08. doi:10.1177/0310057×9902700413. PMID 10470398.
- ^ Yasui R, Culclasure T, Kaufman D, Freed C (1997). “Flecainide overdose: is cardiopulmonary support the treatment?”. Annals of Emergency Medicine. 29 (5): 680–82. doi:10.1016/S0196-0644(97)70257-9. PMID 9140253.
- ^ Latini R, Cavalli A, Maggioni AP, Volpi A (December 1987). “Flecainide distribution in human tissues”. British Journal of Clinical Pharmacology. 24 (6): 820–22. doi:10.1111/j.1365-2125.1987.tb03252.x. PMC 1386410. PMID 3125854.
- ^ Ozkan M, Dweik RA, Ahmad M (September 2001). “Drug-induced lung disease”. Cleve Clin J Med. 68 (9): 782–85, 789–95. doi:10.3949/ccjm.68.9.782. PMID 11563482.
- ^ Camus P, Fanton A, Bonniaud P, Camus C, et al. (2004). “Interstitial lung disease induced by drugs and radiation”. Respiration. 71 (4): 301–26. doi:10.1159/000079633. PMID 15316202.
- ^ Pesenti S, Lauque D, Daste G, Boulay V, et al. (2002). “Diffuse Infiltrative Lung Disease Associated with Flecainide”. Respiration. 69 (2): 182–85. doi:10.1159/000056325. PMID 11961436.
- ^ Haas M, Pérault MC, Bonnefoy P, Rodeau F, Caron F (2001). “[Interstitial pneumopathy due to flecainide]”. Presse Med. 30 (21): 1062. PMID 11471279.
- ^ Robain A, Perchet H, Fuhrman C (February 2000). “Flecainide-associated pneumonitis with acute respiratory failure in a patient with the LEOPARD syndrome”. Acta Cardiol. 55 (1): 45–57. doi:10.2143/ac.55.1.2005718. PMID 10707759.
- ^ Smith G (1985). “Flecainide: a new class Ic antidysrhythmic”. Drug Intell Clin Pharm. 19(10): 703–07. doi:10.1177/106002808501901001. PMID 3902429.
- ^ Padrini R, Piovan D, Busa M, al-Bunni M, Maiolino P, Ferrari M (1993). “Pharmacodynamic variability of flecainide assessed by QRS changes”. Clin Pharmacol Ther. 53 (1): 59–64. doi:10.1038/clpt.1993.9. PMID 8422742.
- ^ Haefeli W, Bargetzi M, Follath F, Meyer U (1990). “Potent inhibition of cytochrome P450IID6 (debrisoquin 4-hydroxylase) by flecainide in vitro and in vivo”. J Cardiovasc Pharmacol. 15 (5): 776–79. doi:10.1097/00005344-199005000-00013. PMID 1692938.
- ^ Abdelilah, Ghannam; et al. (2014). “Non-Accidental Flecainide Overdose, A Case Report”. International Journal of Medicine and Surgery. 1 (2): 53. doi:10.15342/ijms.v1i2.18.
- ^ Ramos E, O’leary M (2004). “State-dependent trapping of flecainide in the cardiac sodium channel”. J Physiol. 560 (Pt 1): 37–49. doi:10.1113/jphysiol.2004.065003. PMC 1665201. PMID 15272045.
- ^ Wang Z, Fermini B, Nattel S (1993). “Mechanism of flecainide’s rate-dependent actions on action potential duration in canine atrial tissue”. J Pharmacol Exp Ther. 267 (2): 575–81. PMID 8246130.
- ^ Mehra D, Imtiaz MS, van Helden DF, Knollmann BC, Laver DR (2014). “Multiple modes of ryanodine receptor 2 inhibition by flecainide”. Molecular Pharmacology. 86 (6): 696–706. doi:10.1124/mol.114.094623. PMC 4244595. PMID 25274603.
- ^ Hilliard FA, Steele DS, Laver D, Yang Z, Le Marchand SJ, Chopra N, Piston DW, Huke S, Knollmann BC (2010). “Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass”. Journal of Molecular and Cellular Cardiology. 48 (2): 293–301. doi:10.1016/j.yjmcc.2009.10.005. PMC 2813417. PMID 19835880.
- ^ Smith GL, MacQuaide N (2015). “The direct actions of flecainide on the human cardiac ryanodine receptor: keeping open the debate on the mechanism of action of local anesthetics in CPVT”. Circulation Research. 116 (8): 1284–86. doi:10.1161/CIRCRESAHA.115.306298. PMID 25858058.
External links
 |
|
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /flɛˈkeɪnaɪd/flek-AY-nyde |
| Trade names | Tambocor, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a608040 |
| Pregnancy category |
|
| ATC code | |
| Pharmacokinetic data | |
| Bioavailability | 95% |
| Protein binding | 40% |
| Metabolism | CYP2D6 (limited) |
| Elimination half-life | 20 hours (range 12–27 hours) |
| Excretion | Kidney |
| Identifiers | |
| CAS Number | |
| PubChemCID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard(EPA) | |
| ECHA InfoCard | 100.211.334 |
| Chemical and physical data | |
| Formula | C17H20F6N2O3 |
| Molar mass | 414.343 g/mol g·mol−1 |
| 3D model (JSmol) | |
| Chirality | Racemic mixture |
/////////Flecainide acetate, E-0735, R-818