GDC 0994
GDC-0994; Ravoxertinib; 1453848-26-4; GDC0994; UNII-R6AXV96CRH; R6AXV96CRH, RG7842; RG-7842; RG 7842
CAS 1453848-26-4
1-[(1S)-1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl]-4-[2-[(2-methylpyrazol-3-yl)amino]pyrimidin-4-yl]pyridin-2-one
PHASE 1
Ravoxertinib also known as GDC-0994 and RG7842, is an orally available inhibitor of extracellular signal-regulated kinase (ERK), with potential antineoplastic activity. Upon oral administration, GDC-0994 inhibits both ERK phosphorylation and activation of ERK-mediated signal transduction pathways. This prevents ERK-dependent tumor cell proliferation and survival. The mitogen-activated protein kinase (MAPK)/ERK pathway is upregulated in a variety of tumor cell types and plays a key role in tumor cell proliferation, differentiation and survival.
GDC-0994 is an ERK inhibitor invented by Array under a collaboration agreement with Genentech. Array has received certain clinical milestones and is entitled to additional potential clinical and commercial milestones and royalties on product sales under the agreement. ERK is a key protein kinase in the RAS/RAF/MEK/ERK pathway, which regulates several key cellular activities including proliferation, differentiation, migration, survival and angiogenesis. Inappropriate activation of this pathway has been shown to occur in many cancers. GDC-0994 is currently advancing in a Phase 1 trial in patients with solid tumors.


WO2013130976
- OriginatorArray BioPharma
- DeveloperGenentech
- ClassAntineoplastics; Small molecules
- Mechanism of ActionExtracellular signal-regulated MAP kinase inhibitors; Mitogen activated protein kinase 3 inhibitors; Mitogen-activated protein kinase 1 inhibitors
Most Recent Events
- 29 Nov 2016Pharmacodynamics data from a preclinical trial in Solid tumours presented at the 28th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics (EORTC-NCI-AACR-2016)
- 29 Nov 2016Adverse events, efficacy, pharmacokinetics and pharmacodynamics data from a phase I trial in Solid tumours presented at the 28th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics
- 16 Jul 2016No recent reports of development identified for phase-I development in Solid-tumours(Late-stage disease, Monotherapy, Second-line therapy or greater) in USA
FREE FORM

(S)-1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one,
mp = 304.4 °C,
[α]D23 +113.8 (c 0.29, MeOH);
IR 1660, 1582, 1557 cm–1;
1H NMR (500 MHz, DMSO-d6) δ 9.69–9.52 (s, 1H), 8.69–8.49 (d, J = 5.1 Hz, 1H), 8.09–7.77 (d, J = 7.3 Hz, 1H), 7.61–7.56 (t, J = 8.1 Hz, 1H), 7.50–7.47 (d, J = 5.1 Hz, 1H), 7.46–7.42 (dd, J = 10.6, 2.0 Hz, 1H), 7.38–7.36 (d, J = 1.9 Hz, 1H), 7.22–7.08 (m, 2H), 6.98–6.79 (dd, J = 7.3, 2.1 Hz, 1H), 6.35–6.23 (d, J = 1.9 Hz, 1H), 6.06–5.90 (dd, J = 8.1, 5.4 Hz, 1H), 5.40–5.23 (d, J = 5.2 Hz, 1H), 4.24–3.97 (m, 2H), 3.80–3.56 (s, 3H);
13C NMR (101 MHz, DMSO-d6) δ 162.18, 161.53, 160.88, 160.55, 157.59 (d, JCF = 246.83 Hz), 147.19, 140.09 (d, JCF = 6.47 Hz), 138.30, 137.75, 137.42, 131.20, 125.53 (d, JCF = 3.45 Hz), 119.29 (d, JCF = 17.45 Hz), 117.74, 116.61 (d, JCF = 21.68 Hz), 109.68, 103.34, 99.36, 61.24, 59.20, 35.93.
HRMS (ESI): m/z [M + H]+ calcd for C21H18ClFN6O2, 441.1242; found, 441.1230.
GDC-0994 benzenesulfonate salt

CAS 1817728-45-2, C21 H18 Cl F N6 O2 . C6 H6 O3 S
GDC-0994 as a light yellow solid,
mp 197.7 °C;
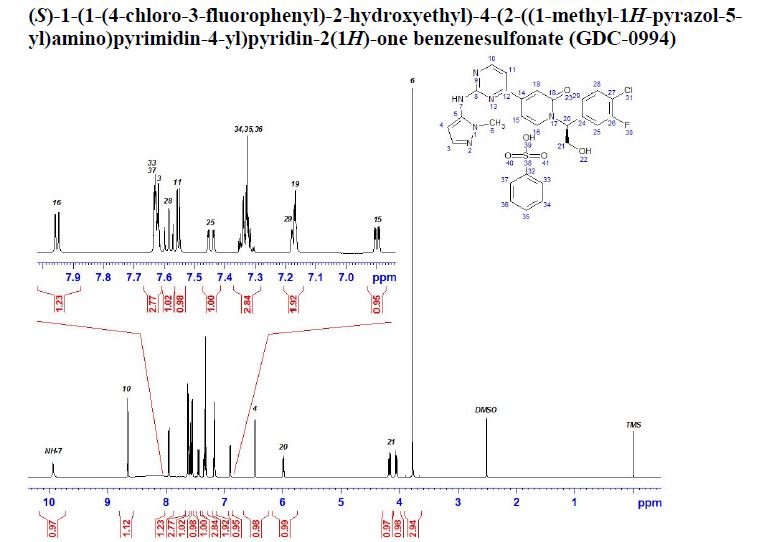
1H NMR (600 MHz, DMSO-d6): 9.93, (s, 1H), 8.65 (d, J = 5.2 Hz, 1H), 7.95 (d, J = 7.27 Hz, 1H), 7.63 (m, 2H), 7.62 (d, J = 1.5 Hz, 1H), 7.58 (t, J = 8.2 Hz, 1H), 7.55 (d, J = 5.2 Hz, 1H), 7.44 (dd, J = 10.6, 1.9 Hz, 1H), 7.33 (m, 3H), 7.18 (d, J = 2.0 Hz, 1H), 7.17 (d, J = 2.1 Hz, 1H), 6.90 (dd, J = 7.3, 2.1 Hz, 1H), 6.48 (d, J = 2.2 Hz, 1H), 5.99 (dd, J = 8.1, 5.5 Hz, 1H), 4.17 (dd, J = 11.9, 8.2 Hz, 1H), 4.05 (dd, J = 11.9, 5.5 Hz, 1H), 3.78 (s, 3H).

13C NMR (150 MHz, DMSO-d6): 161.60, 161.14, 160.02, 159.79, 157.02 (d, J = 245 Hz), 148.0, 146.49, 139.53 (d, J = 6.0 Hz), 139.04, 136.96, 136.39, 130.66, 128.42, 127.59, 125.38, 124.99 (d, J = 3.0 Hz), 118.72 (d, J = 18.0 Hz), 117.29, 116.05 (d, J = 22.5 Hz), 109.75, 102.79, 98.77, 60.64, 58.68, 35.29.
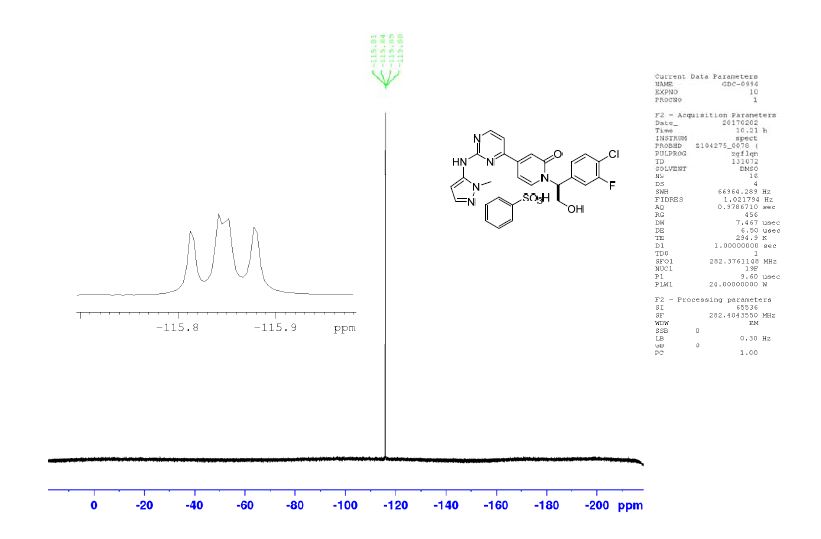
19F NMR (282 MHz, DMSO-d6) −115.86 (dd, J = 10.6, 7.8).
HRMS calcd for C21H18ClFN6O2 [M + H] 441.1242, found 441.1245.
PATENT
WO 2013130976
Example 39

(S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5- yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one
[00398] Step A: (S)-1-(2-(tert-Butyldimethylsiloxy)-1-(4-chloro-3-fluorophenyl)ethyl)-4-(2-(methylsulfonyl)pyrimidin-4-yl)pyridine-2(1H)-one (47 mg, 0.087 mmol), 2-methyl pyrazole-3 -amine (0.175 mmol, 2.0 equivalents) and anhydrous DMF (3.0 mL) were added to a 25 mL round bottomed flask equipped with a stirring bar. The flask was capped with a rubber septum and flushed with nitrogen. Under a blanket of nitrogen, sodium hydride (8.5 mg, 60% dispersion in mineral oil) was added in one portion. The flask was flushed with
nitrogen, capped and stirred at room temperature. The reaction progress was monitored by LCMS, and after 30 minutes, the starting material was consumed. The reaction mixture was quenched by the addition of water (0.5 mL) and ethyl acetate (15 mL). The contents of the round bottomed flask were transferred to a 125 mL separatory funnel, and the reaction flask was rinsed several times with additional ethyl acetate. Crude (S)-1-(2-((tert-butyldimethylsilyl)oxy)-1-(4-chloro-3-fluorophenyl)ethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one was partitioned between ethyl acetate and water (80 mL/30 mL). The ethyl acetate layer was washed once with brine, dried (MgSO4), filtered and concentrated to give crude (S)-1-(2-((tert-butyldimethylsilyl)oxy)-1-(4-chloro-3-fluorophenyl)ethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one. The crude was taken directly into the deprotection step.
[00399] Step B: Crude (S)-1-(2-((tert-butyldimethylsilyl)oxy)-1-(4-chloro-3-fluorophenyl)ethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (48 mg) was dissolved in ethyl acetate (4 mL) and treated dropwise slowly (over 2 minutes) with an ethyl acetate solution (1.0 mL, which had been saturated with HCl gas). The reaction stirred at room temperature for 15 minutes, after which time LCMS indicated complete consumption of the starting material. The reaction mixture was concentrated to an oily residue and purified by prep RP HPLC to yield (S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (20.8 mg, 54.6% yield) as a lyophilized powder. 1H NMR (400 MHz, (CD3)2SO) δ 9.58 (s, 1H), 8.60 (d, J = 5.1 Hz, 1H), 7.91 (t, J = 9.0 Hz, 1H),7.58 (t, J = 8.1 Hz, 1H), 7.52-7.41 (m, 2H), 7.37 (d, J = 1.8 Hz, 1H), 7.14 (dd, J = 10.7,5.1 Hz 2H), 6.86 (dd, J = 7.3, 1.8 Hz, 1H), 6.27(d, J = 1.7 Hz, 1H), 5.97 (dd, J = 7.7, 5.7 Hz, 1H), 5.31(t, J = 5.2 Hz, 1H), 4.15 (m, 1H), 4.10-3.95 (m,1H), 3.69 (s, 3H); LCMS m/z 441 (M+H)+.
PAPER
Discovery of (S)-1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (GDC-0994), an Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) Inhibitor in Early Clinical Development
James F. Blake†, Michael Burkard†, Jocelyn Chan‡, Huifen Chen‡, Kang-Jye Chou‡, Dolores Diaz‡, Danette A. Dudley‡, John J. Gaudino†, Stephen E. Gould‡, Jonas Grina†, Thomas Hunsaker‡, Lichuan Liu‡, Matthew Martinson†, David Moreno†, Lars Mueller‡, Christine Orr‡, Patricia Pacheco‡, Ann Qin‡, Kevin Rasor†, Li Ren†, Kirk Robarge‡, Sheerin Shahidi-Latham‡, Jeffrey Stults‡, Francis Sullivan†, Weiru Wang‡, Jianping Yin‡, Aihe Zhou‡, Marcia Belvin‡, Mark Merchant‡, John Moffat‡, and Jacob B. Schwarz*‡
† Array BioPharma Inc., 3200 Walnut Street, Boulder, Colorado 80301, United States
‡ Genentech, Inc., 1 DNA Way, South San Francisco, California 94080, United States
J. Med. Chem., 2016, 59 (12), pp 5650–5660
DOI: 10.1021/acs.jmedchem.6b00389
The extracellular signal-regulated kinases ERK1/2 represent an essential node within the RAS/RAF/MEK/ERK signaling cascade that is commonly activated by oncogenic mutations in BRAF or RAS or by upstream oncogenic signaling. While targeting upstream nodes with RAF and MEK inhibitors has proven effective clinically, resistance frequently develops through reactivation of the pathway. Simultaneous targeting of multiple nodes in the pathway, such as MEK and ERK, offers the prospect of enhanced efficacy as well as reduced potential for acquired resistance. Described herein is the discovery and characterization of GDC-0994 (22), an orally bioavailable small molecule inhibitor selective for ERK kinase activity.
PATENT
WO 2015154674
https://www.google.com/patents/WO2015154674A1?cl=pt
The present invention provides processes for the manufacture of I which is a useful intermediate that can be used in the manufacture VIII. (WO2013/130976) Compound VIII is an ERK inhibitor and a useful medicament for treating hyperproliferative disorders. The process provides an efficient route to VIII and to the useful intermediates VI and VII. Alkylation of VII with VI affords I, which ultimately is condensed with 1-methyl-1H-pyrazol-5-amine (XIV) . (SCHEME A)

(i) i-PrMgCl, LiCl, THF; (ii) HCO2Na, HCO2H, H2O, EtOH; (iii) GDH-105, morpholineethanesulfonic acid, MgCl2, PEG6000, heptane, 1wt% KRED-NADH-112, NAD, glucose (iv) TBSCl, DMAP, TEA, DCM, 20-25℃ , 15h; (v) MsCl, DCM, 20-25℃ , 3h
Scheme 1. Original Synthetic Process

Scheme 2. Improved Process To I
[0170]

[0173]
2- ( (tert-Butyldimethylsilyl) oxy) -1- (4-chloro-3-fluorophenyl) ethyl methanesulfonate
[0175]
Step 1: 4-bromo-1-chloro-2-fluorobenzene (64 kg) and dry toluene (170kg) were charged to the 2000 L steel reaction vessel under nitrogen. The reactor was evacuated and backfilled with N2 for three times, and cooled to between-10 and 5 ℃ under nitrogen atmosphere. To the solution was added dropwise i-PrMgCl. LiCl (280kg, 1.3M in THF) at between-10 and 10 ℃ . The reaction was stirred for a further 15 to 30min at between-10 and 10 ℃ and then warmed to about 20 to 25 ℃ over 1h. The reaction mixture was stirred for another 6 h stir to complete the exchange. The resulting solution was cooled to between-50 and-40 ℃ . A solution of 2-chloro-N-methoxy-N-methylacetamide (44.5kg) in dry toluene (289kg) was added dropwise to the above solution at while maintaining the temperature between-50 and-30 ℃ . The reaction mixture was warmed to between 20 and 25 ℃ over 1h and then stirred for 3h to complete the reaction. The reaction was quenched by addition of 1N aq. HCl (808l g) at a temperature between-5 and 15 ℃ . The aqueous layer was separated and organic layer was filtered through a pad of diatomaceous earth. The organic layer was washed with 10%aq. NaCl solution (320kg) twice, then concentrated to about 300L to obtain 1- (4-chloro-3-fluorophenyl) -2-chloroethanone (51.8kg, 81.9%yield) as product in toluene.
[0176]
Step 2: The solution of II (51.7kg) in toluene was concentrated and solvent exchanged to EtOH to afford a suspension of II in EtOH (326kg) . A solution of HCOONa·2H2O (54.8kg) and HCOOH (44.5kg) in water (414kg) was added at a temperature between 15 and 35 ℃ under a nitrogen atmosphere. The resulting mixture was heated to reflux and stirred for 4 to 5 h. The solution was cooled to between 20 and 30 ℃ after over 95%conversion occurred. Water (450kg) was added dropwise at between 10 and 30℃ for over 2 h. The resulting suspension was cooled to between-10 and-3 ℃ and the cooled solution stirred for 1 to 2 h. The solid was filtered and the filter cake washed with water (400 kg) to remove the residual HCOONa and HCOOH. The 1- (4-chloro-3-fluorophenyl) -2-hydroxyethanone obtained was suspended in EtOAc (41kg) and n-heptane (64kg) , then warmed to between 45 and 50 ℃ , stirred for 2h, then cooled to between-2 and 5 ℃ for over 2h and stirred at this temperature for 2h. The solids were filtered and dried in vacuo at between 40 and 50 ℃ for 12 h to afford the product as white solid (40.0kg, 99.3%purity, 84.5%yield) .
[0177]
Step 3: A 500 L reactor under nitrogen was charged with purified water (150 kg) , 4-morpholineethanesulfonic acid (0.90kg) , anhydrous MgCl2(0.030kg) , n-heptane (37kg) , 1- (4-chloro-3-fluorophenyl) -2-hydroxyethanone (30kg) , D- (+) -glucose monohydrate (34.8kg) and PEG 6000 (30.0kg) . The pH of the solution was adjusted to between 6.5 and 7.0 with 1N aq. NaOH at between 28 and 32 ℃ . The cofactor recycling enzyme, glucose dehydrogenase GDH-105 (0.300kg) (Codexis Inc., Redwood City, CA, USA) , the cofactor nicotinamide adenine dinucleotide NAD (0.300kg) (Roche) and the oxidoreductase KRED-NADH-112 (0.300kg) (Codexis Inc., Redwood City, CA, USA) were added. The resulting suspension was stirred at between 29 and 31 ℃ for 10 to 12 h while adjusting the pH to maintain the reaction mixture pH between 6.5 and 7.0 by addition of 1N aq. NaOH (160kg) . The pH of the reaction mixture was adjusted to between 1 and 2 by addition of 49%H2SO4 (20kg) to quench the reaction. EtOAc (271kg) was added and the mixture was stirred at between 20 and 30 ℃for 10-15min then filtered through a pad of diatomaceous earth. The filter cake was washed with EtOAc (122kg) . The combined organic layers were separated and aqueous layer was extracted with EtOAc (150kg) . Water (237kg) was added to the combined organic layers. The pH of the mixture was adjusted to between 7.0 and 8.0 by addition of solid NaHCO3. The organic layer was separated, concentrated and then diluted with DCM to afford (R) -1- (4-chloro-3-fluorophenyl) ethane-1, 2-diol (30.9kg, yield 100%) as product in DCM.
[0178]
Step 4: A 1000 L reactor under nitrogen was charged with (R) -1- (4-chloro-3-fluorophenyl) ethane-1, 2-diol (29.5kg) and dry DCM (390kg) . The solution was cooled to between-5 and 0 ℃ . tert-Butylchlorodimethylsilane (25.1 kg) was added in portions while maintaining the temperature between-5 and 2 ℃ . A solution of DMAP (0.95kg) and TEA (41.0kg) in dry DCM (122kg ) was added dropwise to above solution at between-5 and 2 ℃ . The reaction solution was stirred for 1 h, then warmed to between 20 and 25 ℃ and stirred for 16 h. The solution of (R) -2- ( (tert-butyldimethylsilyl) oxy) -1- (4-chloro-3-fluorophenyl) ethanol was recooled to between-10 and-5 ℃ . A solution of methanesulfonyl chloride (19.55 kg) in dry DCM (122kg) was added dropwise to the above solution of while maintaining the temperature between-10 and 0 ℃ . The reaction solution was stirred at between-10 and 0 ℃ for 20 to 30 min, and then warmed to between 0 and 5℃ for over 1h, and stirred. The reaction solution was washed with water (210kg) , followed by 5%aq. citric acid (210kg) , 2%aq. NaHCO3 (210kg) and finally water (2 x 210kg) . The resulting DCM solution was dried (Na2SO4) , filtered and concentrated in vacuo below 15℃ (jacket temperature below 35℃) to afford (R) -2- ( (tert-butyldimethylsilyl) oxy) -1- (4-chloro-3-fluorophenyl) ethyl methanesulfonate (49.5kg, 83.5%yield, KarlFischer=0.01%) as product in DCM.
[0180]
4- (2- (methylsulfonyl) pyrimidin-4-yl) pyridin-2 (1H) -one
[0182]
Step 1: A 1000 L reactor was charged with 2-fluoro-4-iodopyridine (82.2kg) and dry THF (205 kg) . The reactor was evacuated and backfilled with N2three times then cooled to between-30 and-20 ℃ . To the solution was added dropwise i-PrMgCl·LiCl (319 kg, 1.3M in THF) . The reaction was warmed to between-20 and-10℃ and stirred for 1.5 h to complete the transmetallation.
[0183]
A 2000 L reactor was charged with 4-chloro-2-methylthiopyrimidine (45.6kg) , dry THF (205kg) and [1, 3-bis (2, 6-diisopropylphenyl) imidazol-2-ylidene] (3-chloropyridyl) palladium (II) dichloride (PEPPSITM-IPr, 1.850kg) . The 2000 L reactor was evacuated and backfilled with N2 three times and heated to between 55 and 57 ℃ . To the reactor was added over 0.5 to 1 h, the solution of (2-fluoropyridin-4-yl) magnesium chloride while maintaining the temperature between 50 and 62℃ . The resulting reaction mixture was stirred at between 50 and 62 ℃ for a further 2h. The reaction mixture was cooled to between 5 and 25℃ while the reaction was quenched with water (273kg) . The pH of the mixture was adjusted to 8 to 9 by adding solid citric acid monohydrate (7.3kg) . The organic layer was separated, washed with 12.5%aqNaCl (228kg) and concentrated in vacuo below 50℃ to afford 4- (2-fluoropyridin-4-yl) -2- (methylthio) pyrimidine (38.3kg, 61%yield) as product in THF.
[0184]
Step 2: The solution of 4- (2-fluoropyridin-4-yl) -2- (methylthio) pyrimidine (38.2kg) in THF was concentrated and co-evaporated with THF to remove residual water. The suspension was filtered through a pad of diatomaceous earth to remove inorganic salts. To the resulting solution in THF (510kg) was added tert-BuOK (39.7kg) in portions while maintaining the temperature between 15 and 25 ℃ . The mixture was warmed to between 20 and 25 ℃ and stirred for 5h. NaHCO3 (14.9kg) added charged and then a citric acid solution (5kg) in THF (15kg) was added to adjust the pH to between 8 and 9. Water (230kg) was added. The mixture was filtered and the filter cake was washed with THF (100kg) . The combined THF solutions were washed with 12.5%aqueous NaCl (320kg) and concentrated to about 380L to afford a solution of 4- (2- (tert-butoxy) pyridin-4-yl) -2- (methylthio) pyrimidine in THF.
[0185]
To the THF solution cooled to between 15 and 30 ℃ was added1N H2SO4 aq. solution (311kg) . The mixture was stirred at this temperature for 4h. MTBE (280kg) was charged and the pH of reaction solution was adjusted to 14 with 30%aqueous NaOH (120kg) . The aqueous layer was separated and the organic phase filtered to remove inorganic salts. The obtained aqueous layer was washed with MTBE (2 x 280kg) . 2-MeTHF (1630kg) and i-PrOH (180kg) were added to the aqueous solution. The pH was then adjusted to 8 slowly with conc. HCl (19kg) . An organic layer separated and aqueous layer was extracted with 2-MeTHF (305kg) . The combined 2-MeTHF extracts were washed with water (300kg) and concentrated to about 100L. MTBE (230kg) was added and stirred at 20-30 ℃ for 0.5h. The solid was filtered and slurried in a mixture solvent of 2-MeTHF (68kg) and MTBE (230kg) . The suspension was stirred at 35-50 ℃ for 3h, and then cooled to 0 to 10 ℃ and stirred at a further 2h.
[0186]
The solid was filtered and dried in vacuo at between 50 and 62 ℃ for 20 h to afford product 4- (2- (methylthio) pyrimidin-4-yl) pyridin-2 (1H) -one as brown solid (33.55kg, 89.6%assay, 79.4%yield) .
[0188]
(S) -1- (2- ( (tert-Butyldimethylsilyl) oxy) -1- (4-chloro-3-fluorophenyl) ethyl) -4- (2- (methylthio) pyrimidin-4-yl) pyridin-2 (1H) -one (XI)
[0190]
Step 1: The THF was co-evaporated from the THF solution of 4- (2- (methylthio) pyrimidin-4-yl) pyridin-2 (1H) -one (25.5kg) to remove residual water. Dry bis- (2-methoxyethyl) ether (75kg) was added. A solution of KHMDS (131kg, 1M in THF) was added dropwise while maintaining the temperature between 25 and 40 ℃ . The mixture was heated to between 75 and 80℃ and stirred for 30 to 40 min. The resulting mixture was cooled to between 20 and 30℃ under nitrogen atmosphere. A solution of (R) -2- ( (tert-butyldimethylsilyl) oxy) -1- (4-chloro-3-fluorophenyl) ethyl methanesulfonate (47.6kg) in THF (50kg) was added over 30 to 60 min while maintaining the temperature between 20 and 40℃ . The reaction solution was warmed to between 80 and 85 ℃ and stirred for 7 h. The solution was cooled to between 5 and 15 ℃ and water (155 kg) was added. The pH of the solution was adjusted to 7.5 with 30%aqueous citric acid (30 kg) . EtOAc (460kg) was added and the mixture was stirred for 20 min. The organic layer was separated and washed with 12.5%aqueous NaCl (510kg) . The combined aqueous layers were extracted with EtOAc (115kg) . The ethyl acetate layers were concentrated to about 360L to afford (S) -1- (2- ( (tert-butyldimethylsilyl) oxy) -1- (4-chloro-3-fluorophenyl) ethyl) -4- (2- (methylthio) pyrimidin-4-yl) pyridin-2 (1H) -one (44.6kg, 75.7%yield) as product in EtOAc.
[0191]
Step 2: To a solution of (S) -1- (2- ( (tert-butyldimethylsilyl) oxy) -1- (4-chloro-3-fluorophenyl) ethyl) -4- (2- (methylthio) pyrimidin-4-yl) pyridin-2 (1H) -one (44.6kg) in EtOAc (401kg, 10vol) cooled to between 5 and 10 ℃ was added in portions MCPBA (58kg) . The reaction mixture was added to a solution of NaHCO3 (48.7kg) in water (304kg) at a temperature between10 and-20℃ . A solution of Na2S2O3 (15kg) in water (150 kg) was added dropwise to consume residual MCBPA. The organic layer was separated and aqueous layer was extracted with EtOAc (130kg) . The combined organic layers were washed with water (301 kg) , concentrated and solvent exchanged to DCM to afford (S) -1- (2- ( (tert-butyldimethylsilyl) oxy) -1- (4-chloro-3-fluorophenyl) ethyl) -4- (2- (methylsulfonyl) pyrimidin- 4-yl) pyridin-2 (1H) -one (45.0kg, 94.9%yield) as product in DCM. The DCM solution was concentrated to about 100 L, filtered through a pad of SiO2 (60kg) and eluted with an EtOAc/DCM gradient (0, 25 and 50%EtOAc) . The fractions were combined and concentrated to get the product which was re-slurried with (acetone: n-heptane=1: 3 v/v) four times to afford the final product (31.94kg, 71%yield) .
[0193]
(S) -1- (1- (4-Chloro-3-fluorophenyl) -2-hydroxyethyl) -4- (2- ( (1-methyl-1H-pyrazol-5-yl) amino) pyrimidin-4-yl) pyridin-2 (1H) -one, benzenesulfonate salt (VIIIb)
[0195]
Step 1: A clean 100 L cylindrical reaction vessel was charged with THF (13 kg) then (S) -1- (2- ( (tert-butyldimethylsilyl) oxy) -1- (4-chloro-3-fluorophenyl) ethyl) -4- (2- (methylsulfonyl) pyrimidin-4-yl) pyridin-2 (1H) -one (I, 5 kg) and 1-methyl-1H-pyrazol-5-amine (1.1 kg) were added sequentially with medium agitation followed by THF (18 kg) . The mixture was cooled to-35 ℃ and to the resulting thin slurry was added slowly a THF solution of LiHMDS (17.4 kg, 1.0 M) at a rate that maintained the internal temperature below-25 ℃ . After the addition was completed, the reaction was held between-35 and-25 ℃ for 20 min and monitored by HPLC. If the HPLC result indicated ≤ 98.5%conversion, additional LiHMDS (0.34 kg, 1.0 M, 0.05 mol%) was added slowly at-35 ℃ . The reaction was quenched slowly at the same temperature with H3PO4 solution (4.4 kg of 85%H3PO4and 15 kg of water) and the internal temperature was kept below 30 ℃ . The reaction was diluted with EtOAc (18 kg) and the phases separated, the organic layer was washed with H3PO4 solution (1.1 kg of 85%H3PO4 and 12 kg of water) followed by a second H3PO4wash (0.55 kg of 85%H3PO4and 12 kg of water) . If 1-methyl-1H-pyrazol-5-remained, the organic layer was washed again with H3PO4 solution (0.55 kg of 85%H3PO4 and 12 kg of water) . Finally the organic layer was washed sequentially with water (20 kg) and a NaCl and NaHCO3 solution (2 kg of NaCl, 0.35 kg of NaHCO3and 10 kg of water) . After the phase separation, residue water in organic solution was removed through an azeotropic distillation with EtOAc to ≤ 0.5% (by KF) and then solution was concentrated to 20-30 L under a vacuum below 50 ℃ . The solvent was then swapped to MeOH using 35 kg of MeOH and then concentrated to between 20 and 30 L for the next step.
[0196]
Step 2: To the methanolic (S) -1- (2- ( (tert-butyldimethylsilyl) oxy) -1- (4-chloro-3-fluorophenyl) ethyl) -4- (2- ( (1-methyl-1H-pyrazol-5-yl) amino) pyrimidin-4-yl) pyridin-2 (1H) -one (IX) solution in MeOH was added HCl (10.7 kg, 1.25 M in MeOH) at RT. It was slightly exothermic. After the addition was completed, the reaction was heated to 45 ℃ . If the reaction was incomplete after 14 to 16 h, additional HCl (1 kg, 1.25 M in MeOH) was added and agitation at 45 ℃ was continued for 2 h. The reaction was equipped with a distillation setup with acid scrubber. The reaction was concentrated to between 20 and 30 L under a vacuum below 50 ℃ . To the resulting solution was added MeOH (35 kg) and the reaction was concentrated to 20 to 30 L again under a vacuum below 50 ℃ . The solvent was then switched to EtOAc using 40 kg of EtOAc. The solvent ratio was monitored by Headspace GC and the solvent swap continued until it was less than 1/5. The solution was concentrated to between 20 and 30 L under a vacuum below 50 ℃ . After the solution was cooled below 30 ℃ , aqueous NaHCO3 (1.2 kg of NaHCO3 and 20 kg of water) was added slowly with a medium agitation and followed by EtOAc (40 kg) . The organic layer was washed with water (2 x 10 kg) then concentrated to 20-30 L under a vacuum below 50 ℃ . The solvent was then switched to MEK using 35 kg of MEK. The residue MeOH was monitored by Headspace GC and the solvent swap continued until the MeOH was < 0.3%. The solution containing (S) -1- (1- (4-chloro-3-fluorophenyl) -2-hydroxyethyl) -4- (2- ( (1-methyl-1H-pyrazol-5-yl) amino) pyrimidin-4-yl) pyridin-2 (1H) -one (VIII) was concentrated to 20 to 30 L under a vacuum below 50 ℃ for the next step.
[0197]
Step 3: The solution of VIII in MEK was transferred to a second 100 L cylindrical reaction vessel through a 1μm line filter. In a separate container was prepared benezenesulfonic acid solution (1.3 kg of benzenesulfonic acid, 1.4 kg of water and 4.4 kg of MEK) . The filtered VIII solution was heated to 75 ℃ and to the resulting solution was added 0.7 kg of the benzenesulfonic acid solution through a 1μm line filter. The clear solution was seeded with crystalline benzenesulfonic acid salt of VIII (0.425 kg) as a slurry in MEK (0.025 kg of VIIIb crystalline seed and 0.4 kg of MEK) which produced a thin slurry. The remaining benzenesulfonic acid solution was then added through a 1μm line filter in 2 h. After addition, the slurry was heated at 75 ℃ for additional 1 h and then cooled to 18 ℃ in a minimum of 3 h. The resulting thick slurry was agitated at 20 ℃ for 14 to 16 h. The solid was filtered using an Aurora dryer. The mother liquor was assayed by HPLC (about . 3%loss) . The solid was then washed with 1μm line filtered 15.8 kg of MEK and water solution (0.8 kg of water and 15 kg of MEK) and followed by 1μm line filtered 30 kg of MEK. Washes were assayed by HPLC (<1%loss) . The wet cake was dried under a vacuum and a nitrogen sweep at a jacket temperature of 45 ℃ for a minimum 12 h to afford the benzenesulfonic acid salt of VIII, which is labeled VIIIb.
[0198]
Additional Examples
[0201]
To a clean 100 L cylindrical reaction vessel was charged 13 kg of THF first. With a medium agitation, 5.0 kg of I and 1.1 kg of 1-methyl-1H-pyrazol-5-amine was charged sequentially and followed by the rest of THF (18 kg) . At-35 ℃ to the resulting thin slurry was added 17.4 kg of LiHMDS (1.0 mol/L) in THF slowly and the internal temperature was remained below-25 ℃ . After addition, the reaction was held between-35 and-25 ℃ for 20 min. The reaction was monitored by HPLC. If the HPLC result indicated ≤ 98.5%conversion, additional 0.34 kg (0.05 mol%) of LiHMDS (1.0 mol/L) in THF was charged slowly at-35 ℃ . Otherwise, the reaction was quenched at the same temperature with 19.4 kg of H3PO4 solution (4.4 kg of 85%H3PO4and 15 kg of water) slowly and the internal temperature was remained below 30 ℃ . The reaction was diluted with 18 kg of EtOAc. After the phase separation, the organic layer was washed with 13.1 kg of H3PO4 solution (1.1 kg of 85%H3PO4 and 12 kg of water) and then with 12.6 kg of H3PO4solution (0.55 kg of 85%H3PO4 and 12 kg of water) . The organic layer was assayed for the 1-methyl-1H-pyrazol-5-amine level by HPLC. If the HPLC result indicated ≥ 20 μg/mL of 1-methyl-1H-pyrazol-5-amine, the organic layer needed an additional wash with 12.6 kg of H3PO4 solution (0.55 kg of 85%H3PO4 and 12 kg of water) . Otherwise, the organic layer was washed with 20 kg of water. The organic layer was assayed again for the 1-methyl-1H-pyrazol-5-amine level. If the HPLC result indicated ≥ 2 μg/mL of 1-methyl-1H-pyrazol-5-amine, the organic layer needed an additional wash with 20 kg of water. Otherwise, the organic layer was washed with 12.4 kg of NaCl and NaHCO3 solution (2 kg of NaCl, 0.35 kg of NaHCO3 and 10 kg of water) . After the phase separation, residue water in organic solution was removed through an azeotropic distillation with EtOAc to ≤ 0.5% (by KF) and then the solution was concentrated to 20 to 30 L under a vacuum below 50 ℃ . The solvent was then swapped to MeOH using 35 kg of MeOH and then concentrated to 20 to 30 L for the next step.
[0204]
To the IX solution in MeOH from the last step was charged 10.7 kg of HCl (1.25 M in MeOH) at the ambient temperature. It was observed slightly exothermic. After addition, the reaction was heated to 45 ℃ . After 14-16 h, the reaction was monitored by HPLC. If the HPLC result indicated the conversion was ≤ 98%, an additional 1 kg of HCl (1.25 M in MeOH) was charged and the reaction was agitated at 45 ℃ for additional 2 h. Otherwise, the reaction was equipped with a distillation setup with acid scrubber. The reaction was concentrated to 20 to 30 L under a vacuum below 50 ℃ . To the resulting solution was charged 35 kg of MeOH and the reaction was concentrated to 20 to 30 L again under a vacuum below 50 ℃ . The solvent was then switched to EtOAc using 40 kg of EtOAc. The solvent ratio was monitored by Headspace GC. If the ratio of MeOH/EtOAc was greater than 1/5, the solvent swap should be continued. Otherwise, the solution was concentrated to 20 to 30 L under a vacuum below 50 ℃ . After the solution was cooled below 30 ℃ , 21.2 kg of NaHCO3 solution (1.2 kg of NaHCO3 and 20 kg of water) was charged slowly with a medium agitation and followed by 40 kg of EtOAc. After the phase separation, the organic layer was washed with 2 X 10 kg of water. The organic layer was concentrated to 20 to 30 L under a vacuum below 50 ℃ . The solvent was then switched to MEK using 35 kg of MEK. The residue MeOH was monitored by Headspace GC. If the level of MeOH was ≥ 0.3%, the solvent swap should be continued. Otherwise, the solution was concentrated to 20 to 30 L under a vacuum below 50 ℃ for the next step.
[0207]
The VIII solution in MEK from the last step was transferred to a second 100 L cylindrical reaction vessel through a 3 μm line filter. In a separated container was prepared 7.1 kg of benzenesulfonic acid solution (1.3 kg of benzenesulfonic acid, 1.4 kg of water and 4.4 kg of MEK) . The filtered G02584994 solution was heated to 75 ℃ and to the resulting solution was charged 0.7 kg of benzenesulfonic acid solution (10%) through a 3 μm line filter. To the clear solution was charged 0.425 kg of VIIIb crystalline seed slurry in MEK (0.025 kg of VIIIb crystalline seed and 0.4 kg of MEK) . This resulted in a thin slurry. The rest of benzenesulfonic acid solution was then charged through a 3 μm line filter in 2 h. After addition, the slurry was heated at 75 ℃ for additional 1 h and then cooled to 20 ℃ in a minimum of 3 h. The resulting thick slurry was agitated at 20 ℃ for 14-16 h. Solid was filtered using a filter dryer. Mother liquor was assayed by HPLC (about 3%loss) . Solid was then washed with 3 μm line filtered 15.8 kg of MEK and water solution (0.8 kg of water and 15 kg of MEK) and followed by 3 μm line filtered 30 kg of MEK. Washes were assayed by HPLC (<1%loss) . The wet cake was dried under a vacuum and the nitrogen sweep at a jacket temperature of 45 ℃ for a minimum 12 h.
[0210]
To a clean 100 L cylindrical reaction vessel was charged 16 kg of EtOH first. With a medium agitation, 3.5 kg of VIIIb was charged and then followed by the rest of EtOH (8.5 kg) . The thick slurry was heated to 78 ℃ and water (~1.1 kg) was charge until a clear solution was obtained. The hot solution was filtered through a 3 μm line filter to a second clean 100 L cylindrical reaction vessel. The temperature dropped to 55-60 ℃ and the solution remained clear. To the resulting solution was charged with 0.298 kg of VIIIb crystalline seed slurry in EtOH (0.018 kg of VIIIb crystalline seed and 0.28 kg of EtOH) . The thick slurry was concentrated to 20 to 30 L at 60 ℃ under a vacuum and then cooled 20 ℃ in 3 h. The resulting slurry was agitated at 20 ℃ for 14 to 16 h. Solid was filtered using a filter dryer. The mother liquor was assayed by HPLC (about 10%loss) . Solid was then washed with 3 μm line filtered 11.1 kg of EtOH and water solution (0.56 kg of water and 11 kg of EtOH) and followed by 3 μm line filtered 21 kg of MEK. Washes were assayed by HPLC (3%loss) . The wet cake was dried under a vacuum and the nitrogen sweep at a jacket temperature of 45 ℃ for a minimum 12 h.
[0211]
An additional synthetic process is set forth below.
[0214]
To a clean 100 L cylindrical reaction vessel was charged 18 kg of THF first. With a medium agitation, 4.2 kg of I and 0.91 kg of 1-methyl-1H-pyrazol-5-amine was charged sequentially and followed by the rest of THF (21 kg) . At-40 ℃ to the resulting thin slurry was added 14.9 kg of LiHMDS (1.0 mol/L) in THF slowly and the internal temperature was remained below-30 ℃ . After addition, the reaction was held between-35 and-40 ℃ for 20 min. The reaction was monitored by HPLC. The HPLC result indicated 99.1%conversion. The reaction was quenched at the same temperature with 16.7 kg of H3PO4 solution (3.7 kg of 85%H3PO4 and 13 kg of water) slowly and the internal temperature was remained below 30 ℃ . The reaction was diluted with 17 kg of EtOAc. After the phase separation, the organic layer was washed with 13.1 kg of H3PO4 solution (1.1 kg of 85%H3PO4 and 12 kg of water) and then with 10.5 kg of H3PO4 solution (0.46 kg of 85%H3PO4 and 10 kg of water) . The organic layer was assayed for the 1-methyl-1H-pyrazol-5-amine level by HPLC. The HPLC result indicated 2 μg/mL of 1-methyl-1H-pyrazol-5-amine. The organic layer was washed with 15.8 kg of NaCl solution (0.3 kg of NaCl and 15.5 kg of water) . The organic layer was assayed again for the G02586778 level. The HPLC result indicated 0.5 μg/mL of 1-methyl-1H-pyrazol-5-amine. The organic layer was washed with 10.3 kg of NaCl and NaHCO3 solution (1.7 kg of NaCl, 0.6 kg of NaHCO3and 8 kg of water) . After the phase separation, residue water in organic solution was removed through an azeotropic distillation with EtOAc to ≤ 0.5% (by KF) and then the solution was concentrated to 20 to 30 L under a vacuum below 50 ℃ . The solvent was then swapped to MeOH using 30 kg of MeOH and then concentrated to 20 to 30 L for the next step.
[0217]
To the IX solution in MeOH from the last step was charged 9.0 kg of HCl (1.25 M in MeOH) at the ambient temperature. It was observed slightly exothermic. After addition, the reaction was heated to 45 ℃ . After 16 h, the reaction was monitored by HPLC. The HPLC result indicated the conversion was 99.4%. The reaction was equipped with a distillation setup. The reaction was concentrated to 20 L under a vacuum below 50 ℃ . To the resulting solution was charged 35 kg of MeOH and the reaction was concentrated to 20 L again under a vacuum below 50 ℃ . The solvent was then switched to EtOAc using 40 kg of EtOAc. The solvent ratio was monitored by Headspace GC. If the ratio of MeOH/EtOAc was greater than 1/5, the solvent swap should be continued. Otherwise, the solution was concentrated to 20 L under a vacuum below 50 ℃ . After the solution was cooled below 30 ℃ , 18 kg of NaHCO3 solution (1 kg of NaHCO3 and 17 kg of water) was charged slowly with a medium agitation and followed by 34 kg of EtOAc. After the phase separation, the organic layer was washed with 2 X 8 kg of water. The organic layer was concentrated to 20 L under a vacuum below 50 ℃ . The solvent was then switched to MEK using 35 kg of MEK. The residue MeOH was monitored by Headspace GC. If the level of MeOH was ≥ 0.3%, the solvent swap should be continued. Otherwise, the solution was concentrated to 20 L under a vacuum below 50 ℃ for the next step.
The VIII solution in MEK from the last step was transferred to a second 100 L cylindrical reaction vessel through a 1 μm polish filter. In a separated container was prepared 6.0 kg of benzenesulfonic acid solution (1.1 kg of benzenesulfonic acid, 1.2 kg of water and 3.7 kg of MEK) . The filtered solution was heated to 75 ℃ and to the resulting solution was charged 0.6 kg of benzenesulfonic acid solution (10%) through a 1 μm line filter. To the clear solution was charged 0.36 kg of VIIIb crystalline seed slurry in MEK (0.021 kg of VIIIb crystalline seed and 0.34 kg of MEK) . This resulted in a thin slurry. The rest of benzenesulfonic acid solution was then charged through a 1 μm line filter in 2 h. After addition, the slurry was heated at 75 ℃ for additional 1 h and then cooled to 18 ℃ in a minimum of 3 h. The resulting thick slurry was agitated at 18 ℃ for 14-16 h. Solid was filtered using an Aurora dryer. Solid was then washed with 1 μm line filtered 8.15 kg of MEK and water solution (0.35 kg of water and 7.8 kg of MEK) and followed by 1 μm line filtered 12 kg of MEK.
To a clean 100 L cylindrical reaction vessel was charged 21 kg of EtOH first. With a medium agitation, 3.5 kg of VIIIb was charged and then followed by the rest of EtOH (9 kg) . The thick slurry was heated to 78 ℃ and water (1.2 kg) was charge until a clear solution was obtained. The hot solution was filtered through a 1 μm line filter to a second clean 100 L cylindrical reaction vessel. The temperature dropped to 69 ℃ and the solution remained clear. To the resulting solution was charged with 0.37 kg of VIIIb crystalline seed slurry in EtOH (0.018 kg of VIIIb crystalline seed and 0.35 kg of EtOH) . The thin slurry was concentrated to 20 L at 60-70 ℃ under a vacuum and then cooled 18 ℃ in 3 h. The resulting slurry was agitated at 18 ℃ for 14-16 h. Solid was filtered using a filter dryer. Solid was then washed with 1 μm line filtered 8.6 kg of EtOH and water solution (0.4 kg of water and 8.2 kg of EtOH) . The solution was introduced in two equal portions. The solid was then washed by 1 μm line filtered 6.7 kg of MEK. The wet cake was dried under a vacuum and the nitrogen sweep at a jacket temperature of 35-40 ℃ for a minimum 12 h.
Alternative Synthetic Route (Steps 1 to 10 below)

[0229]
1. Charge compound 1 and MeBrPPh3 to a four-necked jacketed flask with a paddle stirrer under N2
[0230]
2. Charge THF (5.0V., KF<0.02%) to the flask (Note: V is the volume of solution to mass of limited reagent or L/Kg)
[0231]
3. Stir the suspension at 0 ℃
[0232]
4. Add the NaH (60%suspended in mineral oil) portionwise to the flask at 0 ℃
[0233]
5. Stir at 0 ℃ for 30min
[0234]
6. Heat to 30 ℃ and stir for 6 hrs
[0236]
8. Charge PE (petroleum ether) (5.0V. ) to the flask
[0237]
9. Add the crystal seed of TPPO (triphenylphospine oxide) (1 to about 5%wt of total TPPO) to the flask
[0238]
10. Stir at-10 ℃ for 2hrs
[0239]
11. Filter, and wash the cake with PE (5.0V. )
[0240]
12. Concentrate the filtrate to dryness
[0241]
13. Purification of the product by distillation under reduced pressure affords 2 as colorless oil
[0245]
1. Add (DHQD) 2PHAL, Na2CO3, K2Fe (CN) 6, K2OsO2 (OH) 4 into a flask under N2 (Ad-mix beta, Aldrich, St. Louis, MO) .
[0247]
3. Add tBuOH (5V) and H2O (5V)
[0249]
5. Stir the mixture at 0 ℃ for 6h
[0251]
7. Add Na2SO3 to quench the reaction
[0252]
8. Stir at 0 ℃ for 2h
[0253]
9. Filter and wash the cake with EA (ethyl acetate)
[0254]
10. Separate the organic layer
[0255]
11. Filter and concentrate to dryness
[0259]
1. Add IV (1 eq. ) and DCM (5V) to a flask under N2
[0261]
3. Add DMAP (0.1 eq. ) , then TEA (1.5 eq. )
[0262]
4. Add TBSCl (1.05 eq. ) dropwise at 0 ℃
[0263]
5. Stir the mixture at 0 ℃ for 1h
[0264]
6. Add water to quench the reaction
[0265]
7. Separate the layers
[0266]
8. Dry the organic layer over Na2SO4
[0268]
10. Concentrate the filtrate to dryness
[0269]
11. Use for next step directly
[0273]
1. Add V (1.0 eq. ) and DCM (5V) into a flask under N2.
[0275]
3. Add TEA (1.51 eq. )
[0276]
4. Add MsCl (1.05 eq. ) dropwise at 0 ℃
[0277]
5. Stir the mixture at rt for 1h
[0278]
6. Add DCM to dilute the mixture for better stirring
[0279]
7. Add water to quench the reaction
[0280]
8. Separate the layers
[0281]
9. Wash the organic layer with NaHCO3
[0282]
10. Dry over Na2SO4
[0283]
11. Filter and concentrate the filtrate to dryness
[0284]
12. Used for next step directly
[0288]
1. Add VII (1eq. ) and DGME (20V) into flask under N2
[0290]
3. Add KHMDS (1M in THF, 1 eq. )
[0291]
4. Add VI (1.2-1.5 eq. ) in DGME solution
[0292]
5. Stir at 0 ℃ for 5min
[0293]
6. Heat to reflux (jacket 120 ℃) and stir for over 4h
[0295]
8. Quench with water and extraction with MTBE
[0296]
9. Wash with 20%NaCl
[0297]
10. Dry over Na2SO4
[0298]
11. Concentrate to dryness and use to next step directly
[0302]
1. Charge XI (1eq. ) , DCM (8V) into flask under N2
[0303]
2. Add mCPBA by portions
[0304]
3. Stir at room temperature for 2h
[0305]
4. Add 7%NaHCO3 aq. to wash
[0306]
5. Quench with Na2S2O4 aq.
[0307]
6. Wash with 20%NaCl aq.
[0308]
7. Dry over Na2SO4
[0309]
8. Filter and concentrate to dryness
[0310]
9. Slurry the result in MTBE (3V) to afford I
[0314]
1. Add I (1eq. ) , 1-methyl-1H-pyrazol-5-amine (4 eq. ) , Cs2CO3, DMF (4V) into a flask under N2
[0315]
2. Stir at room temperature for 3h
[0316]
3. Work-up to afford product.
[0320]
1. IX was dissolved in MeOH
[0321]
2. HCl (1.25 M in MeOH) was charged at the ambient temperature.
[0322]
3. After addition, the reaction was heated to 45 ℃ for 16 h.
[0323]
4. The reaction was cooled to rt and quenched with aqueous NaHCO3 and diluted with EtOAc
[0324]
5. After the phase separation, the organic layer was washed with water. The organic layer was concentrated to afford the crude VIII
[0328]
1. Charge compound 6-2, XIII, Pd-catalyst and sodium bicarbonate to a four-necked jacketed flask with paddle stirrer under N2
[0329]
2. Charge water and 1, 4-dioxane (5.0V., KF<0.02%) to the flask
[0330]
3. Stir the suspension at 85 ℃ for 16hrs
[0331]
4. Filter through the silica-gel (2.0 X) and diatomaceous earth (0.5X)
[0332]
5. Remove the 1, 4-dioxane by distillation under a vacuum
[0333]
6. Partition between water (2.0V) and EtOAc (5.0V)
[0334]
7. Separate the organic phase and concentrate
[0335]
8. Purify by re-crystallization from PE and EtOAc
[0339]
● Add X into a flask
[0340]
● Add 2M HCl (10-15V)
[0341]
● Heat to 100 ℃ and stir for 3h
[0343]
● Neutralize pH to 7 to 8 with 30%NaOH aq.
[0344]
● Extract with THF
[0345]
● Wash with 20%NaCl aq.
[0347]
● Filter and concentrate to dryness
[0348]
Synthesis of Crystalline (S) -1- (1- (4-chloro-3-fluorophenyl) -2-hydroxyethyl) -4- (2- ( (1-methyl-1H-pyrazol-5-yl) amino) pyrimidin-4-yl) pyridin-2 (1H) -one benzenesulfonate salt
[0349]
(S) -1- (1- (4-chloro-3-fluorophenyl) -2-hydroxyethyl) -4- (2- ( (1-methyl-1H-pyrazol-5-yl) amino) pyrimidin-4-yl) pyridin-2 (1H) -one (21.1 mg, 0.048 mmol) was dissolved in MEK (0.5 mL) . Benzenesulfonic acid (Fluka, 98%, 7.8 mg, 0.049 mmol) was dissolved in MEK (0.5 mL) and the resulting solution added drop wise to the free base solution with stirring. Precipitation occurred and the precipitate slowly dissolved as more benzenesulfonic acid solution was added. A small amount of sticky solid remained on the bottom of the vial. The vial contents were sonicated for 10 minutes during which further precipitation occurred. The solid was isolated after centrifugation and vacuum dried at 40 ℃ using house vacuum.
A process for the preparation of a compound of formula VIII, the process comprising the steps of:

(a) contacting 4-bromo-1-chloro-2-fluorobenzene with a metallating agent in an aprotic organic solvent to afford an organomagnesium compound, which is reacted with 2-chloro-N-methoxy-N-methylacetamide to afford 2-chloro-1- (4-chloro-3-fluorophenyl) ethanone (II) ;

(b) contacting II with sodium formate and formic acid in aqueous ethanol to afford 1- (4-chloro-3-fluorophenyl) -2-hydroxyethanone (III)

(c) contacting III with a ketoreductase to afford (R) -1- (4-chloro-3-fluorophenyl) ethane-1, 2-diol (IV) ;

(d) contacting IV with a silyl chloride (R
a)
3SiCl and at least one base in a non-polar aprotic solvent to afford (V) , and subsequently adding sulfonylchloride R
bS (O)
2Cl to afford VI, wherein R
a is independently in each occurrence C
1-6 alkyl or phenyl and R
b is selected from C
1-4 alkyl or phenyl, optionally substituted with 1 to 3 groups independently selected from C
1-3 alkyl, halogen, nitro, cyano, or C
1-3 alkoxy;

(e) contacting 4- (2- (methylsulfonyl) pyrimidin-4-yl) pyridin-2 (1H) -one (VII) with a strong base in an organic solvent and subsequently adding VI to afford XI;

(f) treating XI with an oxidizing agent to afford I;

(g) treating 1-methyl-1H-pyrazol-5-amine with a strong base in an aprotic solvent at reduced temperature and adding the compound of formula I to afford IX; and,

(h) contacting IX with a de-silylating agent to afford VIII.
PAPER
Development of a Practical Synthesis of ERK Inhibitor GDC-0994
† Small Molecule Process Chemistry, Genentech, Inc., 1 DNA Way, South San Francisco, California 94080, United States
‡ Process Research, F. Hoffmann-La Roche AG, Grenzacherstrasse 124, CH-4070 Basel, Switzerland
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00006

The process development of a synthetic route to manufacture ERK inhibitor GDC-0994 on multikilogram scale is reported herein. The API was prepared as the corresponding benzenesulfonate salt in 7 steps and 41% overall yield. The synthetic route features a biocatalytic asymmetric ketone reduction, a regioselective pyridone SN2 reaction, and a safe and scalable tungstate-catalyzed sulfide oxidation. The end-game process involves a telescoped SNAr/desilylation/benzenesulfonate salt formation sequence. Finally, the development of the API crystallization allowed purging of process-related impurities, obtaining >99.5A% HPLC and >99% ee of the target molecule.
| Patent ID |
Patent Title |
Submitted Date |
Granted Date |
| US2016136150 |
COMPOUNDS AND COMPOSITIONS AS INHIBITORS OF MEK |
2015-11-13 |
2016-05-19 |
| US2016122316 |
SERINE/THREONINE KINASE INHIBITORS |
2016-01-12 |
2016-05-05 |
| US2015111869 |
USE OF A COMBINATION OF A MEK INHIBITOR AND AN ERK INHIBITOR FOR TREATMENT OF HYPERPROLIFERATIVE DISEASES |
2014-08-29 |
2015-04-23 |
| US2015051209 |
COMPOUNDS AND COMPOSITIONS AS INHIBITORS OF MEK |
2014-08-05 |
2015-02-19 |
| US2014249127 |
SERINE/THREONINE KINASE INHIBITORS |
2014-02-14 |
2014-09-04 |
| US8697715 |
Serine/threonine kinase inhibitors |
2013-03-01 |
2014-04-15 |
///////////GDC 0994, Ravoxertinib, 1453848-26-4, GDC0994, UNII-R6AXV96CRH, R6AXV96CRH, RG7842, RG-7842, RG 7842, PHASE 1
CN1C(=CC=N1)NC2=NC=CC(=N2)C3=CC(=O)N(C=C3)C(CO)C4=CC(=C(C=C4)Cl)F















 , Nicholas Wong†, Hans Iding‡, Vera Jost‡, Haiming Zhang†, Stefan G. Koenig†, C. Gregory Sowell†, and Francis Gosselin†
, Nicholas Wong†, Hans Iding‡, Vera Jost‡, Haiming Zhang†, Stefan G. Koenig†, C. Gregory Sowell†, and Francis Gosselin†