![(E)-3-[6-[2-(6-Methylpyridin-2-yl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl]-[1,2,4]triazolo[1,5-a]pyridin-5-yl]prop-2-enamide.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=132246793&t=l)
GFH-018
CAS 2169299-67-4
C21 H19 N7 O, 385.42
(E)-3-[6-[2-(6-methylpyridin-2-yl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl]-[1,2,4]triazolo[1,5-a]pyridin-5-yl]prop-2-enamide
2-Propenamide, 3-[6-[5,6-dihydro-2-(6-methyl-2-pyridinyl)-4H-pyrrolo[1,2-b]pyrazol-3-yl][1,2,4]triazolo[1,5-a]pyridin-5-yl]-, (2E)-
GenFleet Therapeutics
Advanced solid tumor; Cancer
TGF-beta Receptor Type-1 (TGFBR1; ALK5; SKR4; TbetaR-I) Inhibitors
Signal Transduction Modulators
GFH-018 , a TGFBR1 inhibitor, being investigated by GenFleet as an oral tablet formulation, for the treatment of cancer, including advanced solid tumors and hepatocellular carcinoma, in March 2019, the company was developing GFH-018 as a class 1 chemical drug in China, with a clinical trial expected to begin in the second half of 2019.




Transforming growth factor-β (TGF-β) is a multifunctional growth factor superfamily with extensive biological activity, involved in early embryonic development, cartilage and bone formation, extracellular matrix synthesis, inflammation, Interstitial fibrosis, regulation of immune and endocrine functions, tumor formation and development.
The TGF-β superfamily consists of a class of structural and functionally related polypeptide growth factors, including TGF-βs (ie, narrowly defined TGF-β), activins (axivins), inhibins, and bone morphogenetic proteins (BMPs). Müllerian inhibitors (mullerian), etc., TGF-β is one of the important members of this family. In mammals, TGF-β mainly exists in three forms of TGF-β1, TGF-β2 and TGF-β3, which are located on different chromosomes, and TGF-β1 accounts for the highest proportion (>90%) in somatic cells. It has the strongest activity, the most functions, and the widest distribution. The newly synthesized TGF-β appears as an inactive precursor consisting of a signal peptide, a latent-associated polypeptide (LAP) and a mature TGF-β. After enzymatic hydrolysis, it forms active TGF-β, and then Receptor binding exerts a biological effect.
TGF-[beta] signaling molecules signal through a transmembrane receptor complex. TGF-β receptor is a transmembrane protein present on the cell surface and is divided into type I receptor (TGF-βRI), type II receptor (TGF-βRII) and type III receptor (TGF-βRIII), of which TGF- βRI is also known as activin receptor-like kinase 5 (ALK5). TGF-βRIII lacks intrinsic activity and is primarily involved in the storage of TGF-β. TGF-βRI and TGF-βRII belong to the serine/threonine kinase family. Type II receptors bind to TGF-β ligands with higher affinity and form heterologous receptor complexes with type I receptors. Phosphorylation of a region rich in glycine and serine residues (GS domain) of the proximal membrane of the receptor initiates an intracellular signal cascade reaction.
Smads are important TGF-β signal transduction and regulatory molecules in cells, which can directly transduce TGF-β signaling from the cell membrane, such as the nucleus. TGF-β/Smads signaling pathway plays an important role in the occurrence and development of tumors. . In TGF-β/Smads signal transduction, activated TGF-β first binds to TGF-βRII on the cell membrane surface to form a heterodimeric complex, and TGF-βRI recognizes and binds to the binary complex.
TGF-βRII phosphorylates serine/threonine in the GS domain of the cytoplasmic domain of TGF-βRI, thereby activating TGF-βRI; activated TGF-βRI further phosphorylates R-Smads (Smad2/Smad3) protein, which in turn Co-Smad (Smad4) binds to a heterotrimeric complex that enters the nucleus and acts synergistically with other co-activators and co-inhibitors to regulate transcription of target genes. . Any change in any part of the TGF-β/Smads signaling pathway leads to abnormalities in the signal transduction pathway.
Current research indicates that in tumor cells, TGF-β can directly affect tumor growth (non-inherent effects of TGF-β signaling), or by inducing epithelial-mesenchymal transition, blocking anti-tumor immune responses, and increasing tumor-associated fibrosis And enhanced angiogenesis indirectly affects tumor growth (the intrinsic effect of TGF-β). At the same time, TGF-β has a strong fibrotic induction, which is an activator of tumor-associated fibroblasts. These fibroblasts are a major source of collagen type I and other fibrotic factors. Induction products of fibroblasts and other fibrotic factors may continue to develop a microenvironment that reduces immune responses, increases drug resistance, and enhances tumor angiogenesis. In addition, TGF-β affects blood vessels during individual development and tumor growth. Raw regeneration. For example, TGF-βRI-deficient mouse embryos show severe vascular development defects, demonstrating that the TGF-β signaling pathway is a key regulator in vascular endothelium and smooth muscle cell development.
In 2013, the FDA awarded Lilly’s small molecule TGF-βRI inhibitor LY2157299 (WO 2002/094833) for the treatment of glioma and liver cancer. LY2157299 is an orphan drug under research, named Galunisertib. Galunisertib inhibits tumor cell invasion and metastasis while inhibiting the infiltration of tumor cells into blood vessels. In the phase 2 clinical trial of patients with liver cancer, about 23% of patients treated with Galunisertib had a decrease in serum alpha-fetoprotein (AFP) levels of more than 20%. Compared with patients without AFP response, these patients had slower tumor progression and longer survival, and increased expression of cadherin in epithelial cells was also observed in these patients, suggesting that Galunisertib can be regulated by inhibiting TGF-β signaling pathway. EMT, thereby inhibiting the progression of liver cancer, the structure of Galunisertib (LY2157299) is shown in formula (II):
Background research and development materials refer to the following documents:
WO2009/009059; WO2007/076127; WO2004/026306; WO2004/072033; WO2002/094833.
PATENT
WO2017215506
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017215506
Preparation of intermediates 1-6:
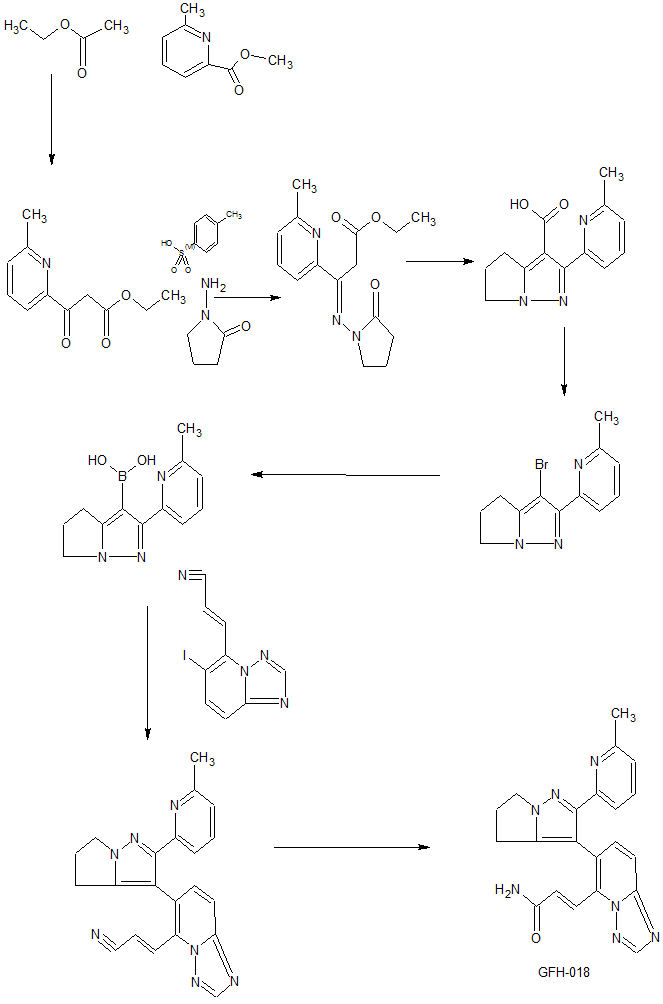
Step A: Ethyl acetate (291.41 ml, 2.98 mol) was dissolved in toluene (750.00 ml), and then sodium ethoxide (135.06 g, 1.98 mol) was added portionwise at room temperature, and the mixture was stirred at room temperature for 1 hour. Methyl 6-methylpyridine-2-carboxylate (150.00 g, 992.33 mmol) was added to the above reaction solution at 25 ° C, then heated to 95 ° C and stirred for 15 hours. The reaction mixture was cooled to 30 ° C, the pH was adjusted to 7 with acetic acid, diluted with water (500 ml), and ethyl acetate (500 ml). The organic phase was dried with anhydrous sodium s The residue was purified with EtOAc EtOAc EtOAc (EtOAc:EtOAc Rate: 58.35%).
Step B: Ethyl 3-(6-methyl-2-pyridine)-3-oxo-propanoate (120.00 g, 579.07 mmol) was dissolved in pyridine (300 mL) then 1-aminopyrrolidine- 2-keto-p-toluenesulfonate (172.01 g, 631.66 mmol). The reaction mixture was stirred at 25 ° C for 16 hours and then concentrated under reduced pressure to remove solvent. The residue was diluted with water (300 ml) and then extracted with ethyl acetate (300 ml). The combined organic phases were dried with anhydrous sodium s , yield: 90.28%).
Step C: Dissolving 3-(6-methyl-2-pyridine)-3-(2-carbonyl-pyrrolidine)imino-propionic acid ethyl ester (155.00 g, 535.72 mmol) in toluene and then adding ethanol Sodium (72.91 g, 1.07 mol). The reaction mixture was heated to 100 ° C and stirred for 16 hours, then cooled to room temperature. It was slowly diluted with water (1.5 liters), adjusted to pH 4 with concentrated hydrochloric acid, and extracted with dichloromethane / isopropyl alcohol (10/1) (1 liter x 7). The combined organic layers were dried with anhydrous sodium s The residue was triturated with petroleum ether / ethyl acetate = 10/1 (200 mL). The solid was dried under reduced pressure to give 2-(6-methyl-2-pyridine)-5,6-dihydro-4H-pyrrole[1,2-b]pyrazole-3-carboxylic acid (52.80 g, yield : 40.52%).
Step D: Dissolving 2-(6-methyl-2-pyridyl)-5,6-dihydro-4H-pyrrole[1,2-b]pyrazole-3-carboxylic acid (45.00 g, 184.99 mmol) In N,N-dimethylformamide (650.00 ml), then NBS (49.09 g, 258.99 mmol). The reaction mixture was stirred at 30-40 ° C for 60 hours, then diluted with water (600 mL) and extracted with dichloromethane / isopropyl alcohol (10/1) (500 mL × 3). The combined organic phases were washed with EtOAc (EtOAc m. The resulting solid was slurried with EtOAc/EtOAc =EtOAc (EtOAc). The solid was dried under reduced pressure to give 3-bromo-2-(6-methyl-2-pyridine)-5,6-dihydro-4H-pyrrole[1,2-b]pyrazole (33.00 g, yield: 64.13%).
Step E: 3-Bromo-2-(6-methyl-2-pyridyl)-5,6-dihydro-4H-pyrrole [1,2-b]pyrazole (1.00 g, 3.60 mmol) and boric acid Triisopropyl ester (1.79 g, 9.54 mmol) was dissolved in tetrahydrofuran (20.00 mL). The reaction mixture was cooled to minus 70 ° C, then n-butyllithium (2.5 M, 3.74 mL) was then added dropwise. After completion of the dropwise addition, the reaction mixture was stirred at 25 ° C for 1 hour, and then the pH was adjusted to 7 with aqueous hydrochloric acid (0.5 mol / liter). The tetrahydrofuran was then concentrated under reduced pressure and cooled to 15 °C. The mixture was filtered, and the filtered cake was purified with EtOAc EtOAc EtOAc (EtOAc) 5,6-Dihydro-4H-pyrrole[1,2-b]pyrazol-3-yl]boronic acid (750 mg, yield: 85.71%).
Preparation of Example 1:
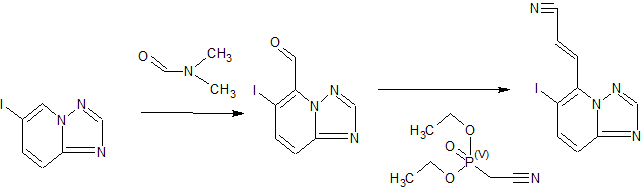
Step A: 6-Iodo-[1,2,4]triazolo[1,5-a]pyridine (16.00 g, 65.30 mmol) was dissolved in tetrahydrofuran (800.00 mL) and cooled to below 60-70 ° C. Thereafter, lithium hexamethyldisilazide (1 mol/liter, 130.60 ml, 65.30 mmol) was added dropwise. The reaction mixture was stirred at minus 60-70 ° C for 15 minutes and N,N-dimethylformamide (14.32 g, 195.90 mmol, 15.07 mL). Stirring was then continued at minus 60 to 70 degrees C for 15 minutes and then quenched with saturated aqueous ammonium chloride (500 mL). The reaction mixture was warmed to room temperature and then extracted with ethyl acetate (500 ml). The combined organic layers were washed with EtOAc EtOAc m. The residue was purified with a silica gel column (eluent: methylene chloride / ethyl acetate = 10/1) to afford 6-iodo-[1,2,4]triazolo[1,5-a]pyridine-5- Formaldehyde (6.40 g, yield: 35.90%). . 1H NMR (400 MHz, DMSO-d6) 10.46 (S, IH), 8.62 (S, IH), 8.16 (D, J = 9.3Hz, IH), 7.88 (D, J = 9.3Hz, IH).
Step B: To a 500 ml three-necked flask equipped with a thermometer and a nitrogen balloon, 2-diethoxyphosphorylacetonitrile (3.83 g, 21.61 mmol, 3.48 ml) and tetrahydrofuran (80 ml) were added. The mixture was cooled to 0.degree. C. and then potassium tert-butoxide (2.42 g, 21.61 mmol). The reaction mixture was stirred at 0 ° C for 15 minutes and then added dropwise to another suspension through a dropping funnel (dispersing 6-iodo-[1,2,4]triazolo[1,5-a]pyridine-5-carbaldehyde In tetrahydrofuran (120 ml) and cooled to 0 ° C). The reaction mixture was stirred at 0<0>C for 15 min then EtOAc (EtOAc)EtOAc. The combined organic layers were washed with EtOAc EtOAc m. The residue was purified with a silica gel column (eluent: methylene chloride / ethyl acetate = 200/1 to 10/1) to afford (E)-3-(6-iodo-[1,2,4]triazole. [1,5-a]pyridin-5-yl)prop-2-enenitrile (4.2 g, yield: 65.66%). . 1 H NMR (400 MHz, CHLOROFORM-D) [delta] 8.42 (S, IH), 8.03 (D, J = 9.3Hz, IH), 7.98-7.91 (m, IH), 7.85-7.78 (m, IH), 7.60 (d, J = 9.2 Hz, 1H).
Step C: (E)-3-(6-Iodo-[1,2,4]triazolo[1,5-a]pyridin-5-yl)prop-2-enenitrile (4.50 g, 15.20 m Mole), [2-(6-methyl-2-pyridyl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl]boronic acid (4.43 g, 18.24 m Mole), sodium carbonate (4.83 g, 45.60 mmol), [1,1′-bis(diphenylphosphino)ferrocene]palladium dichloride (556.07 mg, 759.96 μmol), 2-dicyclohexylphosphine- 2′,6′-dimethoxybiphenyl (311.98 mg, 759.96 μmol) and [2-(2-aminophenyl)phenyl]-chloro-palladium-cyclohexyl-[2-(2,6- Dimethoxyphenyl)phenyl]phosphine (547.64 mg, 759.96 μmol) was added to a mixed solvent of dioxane (100 ml) and water (20 ml). It was replaced with nitrogen 3 times and then heated to 90-100 ° C and stirred for 2 hours. The reaction mixture was poured into water (200 ml) and evaporated and evaporated. The combined organic layers were washed with EtOAc EtOAc m. The residue was purified with EtOAc mjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjj The solid was concentrated and dried under reduced pressure to give (E)-3-[6-[2-(6-methyl-2-pyridyl)-5,6-dihydro-4H-pyrrolo[1,2-b] Pyrazol-3-yl]-[1,2,4]triazolo[1,5-a]pyridin-5-yl]prop-2-enenitrile (5.37 g, yield: 96.16%). . 1 H NMR (400 MHz, CHLOROFORM-D) [delta] 8.49 (S, IH), 7.82-7.74 (m, 2H), 7.59-7.46 (m, 4H), 6.99 (dd, J = 2.6,6.1Hz, IH) , 4.39 (d, J = 6.3 Hz, 2H), 2.90 – 2.70 (m, 4H), 2.20 (s, 3H).
Step D: (E)-3-[6-[2-(6-Methyl-2-pyridyl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole-3 -yl]-[1,2,4]triazolo[1,5-a]pyridin-5-yl]prop-2-enenitrile (5.37 g, 14.62 mmol) dissolved in dichloromethane (20 mL) , a mixed solvent of dimethyl sulfoxide (70 ml) and water (20 ml), then separately added hydrogen peroxide (8.29 g 73.10 mmol, 7.02 ml, 30%) and sodium hydroxide (2 mol / liter, 14.62 ml) ). The mixture was stirred at 15-20 degrees Celsius for 12 hours. The mixture was poured into water (200 ml), and extracted with a mixture solvent of dichloromethane/isopropanol (3/1) (200 ml × 1). The organic layer was washed with EtOAc EtOAc m. The residue was purified by preparative high performance liquid chromatography (column: Phenomenex Gemini C18 250 x 50 mm x 10 μm; mobile phase: [water (0.05% ammonia v/v)-acetonitrile]; gradient: 5%-32%, 33 80% min) Example 1 (3.6 g, yield: 63.82%) was obtained. . 1 H NMR (400 MHz, CHLOROFORM-D) [delta] 8.45 (S, IH), 8.09 (D, J = 15.6Hz, IH), 7.85 (D, J = 15.6Hz, IH), 7.69 (D, J = 9.2 Hz, 1H), 7.55-7.45 (m, 2H), 7.37 (d, J = 7.8 Hz, 1H), 6.99 (d, J = 7.7 Hz, 1H), 5.93-5.65 (m, 2H), 4.35 (br .s., 2H), 2.99-2.64 (m, 4H), 2.33 (s, 3H).
PATENT
WO-2019114792
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019114792&tab=FULLTEXT&maxRec=1000
Novel crystalline and salt (hydrochloride, sulfate and mesylate) forms of a TGF-βRI inhibitor, designated as Forms A and B, processes for their preparation and compositions comprising them are claimed for treating cancers. The compound was originally claimed in WO2017215506 , assigned to Medshine Discovery Inc alone.
Example 1 Preparation of a compound of formula (I)
Preparation of intermediates 1-6:
Step A: Ethyl acetate (291.41 ml, 2.98 mol) was dissolved in toluene (750.00 ml), and then sodium ethoxide (135.06 g, 1.98 mol) was added portionwise at room temperature, and the mixture was stirred at room temperature for 1 hour. 1-1 (150.00 g, 992.33 mmol) was added to the above reaction liquid at 25 ° C, and then heated to 95 ° C and stirred for 15 hours. The reaction mixture was cooled to about 30 ° C, and the pH was adjusted to 7 with acetic acid, diluted with water (500 ml), and ethyl acetate (500 ml). The organic phase was dried with anhydrous sodium s The residue was purified with a silica gel column (eluent: petroleum ether/ethyl acetate v/v = 50/1) to afford 1-2.
Step B: Dissolve 1-2 (120.00 g, 579.07 mmol) in pyridine (300 mL), then add 1-aminopyrrolidin-2-one p-toluenesulfonate (172.01 g, 631.66 mmol) ). The reaction mixture was stirred at 25 ° C for 16 hours and then concentrated under reduced vacuo. The residue was diluted with water (300 ml) and then extracted with ethyl acetate (300 ml). The combined organic layers were dried with anhydrous sodium s
Step C: 1-3 (155.00 g, 535.72 mmol) was dissolved in toluene then sodium ethoxide (72.91 g, 1.07 mol). The reaction mixture was heated to 100 ° C and stirred for 16 hours, then cooled to room temperature. It was slowly diluted with water (1.5 liters), adjusted to pH 4 with concentrated hydrochloric acid, and extracted with dichloromethane/isopropanol (v/v = 10/1, 1 liter x 7). The combined organic layers were dried with anhydrous sodium s The residue was triturated with petroleum ether / ethyl acetate (v/v = 10/1, 200 mL). The solid was dried under reduced pressure to give 1-4.
Step D: 1-4 (45.00 g, 184.99 mmol) was dissolved in N,N-dimethylformamide (650.00 ml), then NBS (49.09 g, 258.99 mmol). The reaction mixture was stirred at 30 to 40 ° C for 60 hours, then diluted with water (600 ml), and extracted with dichloromethane / isopropyl alcohol (v / v = 10 / 1,500 ml × 3). The combined organic phases were washed with EtOAc (EtOAc m. The resulting solid was slurried with EtOAc/EtOAc (EtOAc/EtOAc) The solid was dried under reduced pressure to give 1-5.
Step E: 1-5 (1.00 g, 3.60 mmol) and triisopropyl borate (1.79 g, 9.54 mmol) were dissolved in tetrahydrofuran (20.00 mL). The reaction mixture was cooled to minus 70 ° C, then n-butyllithium (2.5 M, 3.74 mL) was added dropwise. After completion of the dropwise addition, the reaction mixture was stirred at 25 ° C for 1 hour, and then the pH was adjusted to 7 with aqueous hydrochloric acid (0.5 mol / liter). It was then concentrated under reduced pressure to remove tetrahydrofuran and cooled to 15 °C. The mixture was filtered, and the EtOAc EtOAc m.
Preparation of the compound of formula (I):
Step A: 1-7 (16.00 g, 65.30 mmol) was dissolved in tetrahydrofuran (800.00 ml), cooled to minus 60-70 ° C, and lithium hexamethyldisilazide (1 mol/L, 130.60) was added dropwise. ML, 65.30 mmol). The reaction mixture was stirred at -60 to 70 ° C for 15 minutes, and N,N-dimethylformamide (14.32 g, 195.90 mmol, 15.07 ml) was added. Stirring was then continued at minus 60-70 ° C for 15 minutes and then quenched with saturated aqueous ammonium chloride (500 mL). The reaction mixture was warmed to room temperature and then extracted with ethyl acetate (500 ml). The combined organic layers were washed with EtOAc EtOAc m. The residue was purified with a silica gel column (eluent: methylene chloride / ethyl acetate v/v = 10/1) to afford 1-8. . 1 H NMR (400 MHz, DMSO-d6) 10.46 (S, IH), 8.62 (S, IH), 8.16 (D, J = 9.3Hz, IH), 7.88 (D, J = 9.3Hz, IH).
Step B: To a 500 ml three-necked flask equipped with a thermometer and a nitrogen balloon, 2-diethoxyphosphorylacetonitrile (3.83 g, 21.61 mmol, 3.48 ml) and tetrahydrofuran (80 ml) were added. The mixture was cooled to 0 ° C then potassium tert-butoxide (2.42 g, 21.61 mmol). The reaction mixture was stirred at 0<0>C for 15 min then added dropwise to a further suspension (1~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ The reaction mixture was stirred at 0<0>C for 15 min then EtOAc (EtOAc)EtOAc. The combined organic layers were washed with EtOAc EtOAc m. The residue was purified with a silica gel column (eluent: methylene chloride/ethyl acetate v/v = 200/1 to 10/1) to afford 1-9. . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta] 8.42 (S, IH), 8.03 (D, J = 9.3Hz, IH), 7.98-7.91 (m, IH), 7.85-7.78 (m, IH), 7.60 ( d, J = 9.2 Hz, 1H).
Step C: 1-9 (4.50 g, 15.20 mmol), 1-6 (4.43 g, 18.24 mmol), sodium carbonate (4.83 g, 45.60 mmol), [1,1′-bis (diphenyl) Phosphine) ferrocene] palladium dichloride (556.07 mg, 759.96 μmol), 2-biscyclohexylphosphine-2′, 6′-dimethoxybiphenyl (311.98 mg, 759.96 μmol) and [2-( 2-Aminophenyl)phenyl]-chloro-palladium-cyclohexyl-[2-(2,6-dimethoxyphenyl)phenyl]phosphine (547.64 mg, 759.96 μmol) was added to the dioxane (100 ml) and water (20 ml) in a mixed solvent. It was replaced with nitrogen three times and then heated to 90 to 100 ° C and stirred for 2 hours. The reaction mixture was poured into water (200 ml) and evaporated and evaporated. The combined organic layers were washed with EtOAc EtOAc m. The residue was purified on a silica gel column (eluent: methylene chloride/methanol, v/v=30/1) to afford crude crude product in petroleum ether/ethyl acetate (v/v=5/1) After stirring for 12 hours, the solid was collected by filtration, and the solid was concentrated and dried under reduced pressure to give 1-10. . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta] 8.49 (S, IH), 7.82-7.74 (m, 2H), 7.59-7.46 (m, 4H), 6.99 (dd, J = 2.6,6.1Hz, IH), 4.39 (d, J = 6.3 Hz, 2H), 2.90 – 2.70 (m, 4H), 2.20 (s, 3H).
Step D: 1-10 (5.37 g, 14.62 mmol) was dissolved in a mixed solvent of dichloromethane (20 ml), dimethyl sulfoxide (70 ml) and water (20 ml), and then hydrogen peroxide ( 8.29 g 73.10 mmol, 7.02 mL, 30%) and sodium hydroxide (2 mol/L, 14.62 mL). The mixture was stirred at 15 to 20 ° C for 12 hours. The mixture was poured into water (200 ml), and extracted with a mixture solvent of dichloromethane/isopropanol (3/1) (200 ml × 1). The organic layer was washed with EtOAc EtOAc m. The residue was purified by preparative high performance liquid chromatography (column: Phenomenex Gemini C18 250 x 50 mm x 10 μm; mobile phase: [water (0.05% ammonia v/v)-acetonitrile]; gradient: 5%-32%, 33 80% minute) to give a compound of formula (I). . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta] 8.45 (S, IH), 8.09 (D, J = 15.6Hz, IH), 7.85 (D, J = 15.6Hz, IH), 7.69 (D, J = 9.2Hz , 1H), 7.55-7.45 (m, 2H), 7.37 (d, J = 7.8 Hz, 1H), 6.99 (d, J = 7.7 Hz, 1H), 5.93-5.65 (m, 2H), 4.35 (br. s., 2H), 2.99-2.64 (m, 4H), 2.33 (s, 3H).
Example 2 Preparation of a compound of formula (II)
115 mg of the compound of formula (I) was added to an 8 ml glass vial, 4 ml of tetrahydrofuran was added, and the solution was sonicated by ultrasonication; then 1.05 equivalent of p-toluenesulfonic acid monohydrate was slowly added. The suspension sample was placed on a magnetic stirrer (40 ° C) and stirred for 16 hours. The sample solution was centrifuged, and the solid was taken out and dried in a vacuum oven at 35 ° C for 16 hours to obtain a compound of the formula (II). 1 H NMR (400 MHz, CD 3 OD) δ 8.61 (s, 1H), 8.14 (t, J = 8.0 Hz, 1H), 8.05 (d, J = 15.6 Hz, 1H), 7.90 (d, J = 8.8 Hz, 1H), 7.70 (dd, J=8.4, 15.6 Hz, 4H), 7.54 (d, J = 15.6 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 7.6) Hz, 2H), 4.42 (m, 2H), 3.05-2.87 (m, 2H), 2.82 (s, 3H), 2.81-2.74 (m, 2H), 2.35 (s, 3H).
Example 3 Preparation of a compound of formula (IV)
115 mg of the compound of formula (I) was added to an 8 ml glass vial, 4 ml of tetrahydrofuran was added, and the solution was sonicated by ultrasonication; then 1.05 equivalent of hydrochloric acid was slowly added. The suspension sample was placed on a magnetic stirrer (40 ° C) and stirred for 16 hours. The sample solution was centrifuged, and the solid was taken out and dried in a vacuum oven at 35 ° C for 16 hours. The obtained solid was added to an appropriate amount of acetone to prepare a suspension and stirred at 40 ° C, and the supernatant was discarded by centrifugation, and the solid sample was drained with an oil pump at room temperature to obtain a compound of the formula (IV).
Example 4 Preparation of a compound of formula (V)
115 mg of the compound of formula (I) was added to an 8 ml glass vial, 4 ml of tetrahydrofuran was added, and the solution was sonicated by ultrasonication; then 1.05 equivalent of sulfuric acid was slowly added. The suspension sample was placed on a magnetic stirrer (40 ° C) and stirred for 16 hours. The sample solution was centrifuged, and the solid was taken out and dried in a vacuum oven at 35 ° C for 16 hours to obtain a compound of the formula (V).
Example 5 Preparation of a compound of formula (VI)
115 mg of the compound of formula (I) was added to an 8 ml glass vial, 4 ml of tetrahydrofuran was added, and the solution was sonicated by ultrasonication; then 1.05 equivalent of methanesulfonic acid was slowly added. The suspension sample was placed on a magnetic stirrer (40 ° C) and stirred for 16 hours. The sample solution was centrifuged, and the solid was taken out and dried in a vacuum oven at 35 ° C for 16 hours to obtain a compound of the formula (VI).
Example 6 Preparation of Form A of Compound of Formula (I)
10 g of the compound of the formula (I) was placed in a mixed solvent of ethanol (80 ml) and water (40 ml), heated to 70-75 ° C and stirred until clarified, and filtered while hot, and the filtrate was distilled under reduced pressure to a volume of the remaining solution. 50 ml, followed by cooling to stand for crystallisation, filtration, and the resulting filter cake was dried under reduced pressure to give a solid of the compound of formula (I).
Example 7 Preparation of Form B of Compound of Formula (II)
192 mg of the compound of formula (I) was weighed into a glass bottle. 10 ml of a tetrahydrofuran:acetic acid (v/v=9/1) mixed solvent was added, and after ultrasonic assisted for 30 minutes, the sample was dissolved into a clear solution. Stir on a magnetic stirrer (40 ° C). After 1.05 equivalents of p-toluenesulfonic acid monohydrate was slowly added, the sample was stirred overnight. After naturally cooling to room temperature, the supernatant was discarded by centrifugation, stirred for 10 hours by adding 10 ml of tetrahydrofuran, and the supernatant was discarded by centrifugation, and the same procedure was repeated twice more. The obtained solid was dried in a vacuum oven at 40 ° C for 1 hour, and after milling, it was further dried in a vacuum oven at 30 ° C for 16 hours to obtain a crystal form B of the compound of the formula (II).
.///////////////////GFH-018, GFH 018, GenFleet Therapeutics, Advanced solid tumor, Cancer, PRECLINICAL
NC(=O)/C=C/c4n5ncnc5ccc4c2c3CCCn3nc2c1cccc(C)n1