














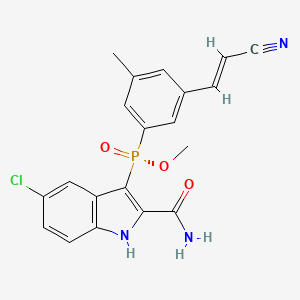
GSK2248761A , IDX899, Fosdevirine,
Fosdevirine; IDX899; IDX-899; GSK2248761; cas 1018450-26-4; GSK-2248761, IDX 12899
| Molecular Formula: | C20H17ClN3O3P |
|---|---|
| Molecular Weight: | 413.798 g/mol |
[R(P)]-(2-Carbamoyl-5-chloro-1H-indol-3-yl)[3-(2-cyanovinyl)-5-methylphenyl]phosphinic acid methyl ester
Phase II clinical trials for the treatment of HIV infection
Idenix (Originator)
Fosdevirine, also known as GSK2248761 and IDX899, a Highly Potent Anti-HIV Non-nucleoside Reverse Transcriptase Inhibitor having an EC50 of 11 nM against the Y181C/K103N double mutant. GSK2248761 is a novel, once-daily (QD), next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI) with activity against efavirenz-resistant strains. GSK2248761 at 100 to 800 mg QD for 7 days was well tolerated, demonstrated potent antiviral activity in treatment-naive HIV-infected subjects, and had favorable PK and resistance profiles. GSK2248761 is no longer in clinical development.
IDX-12899 is a non-nucleoside reverse transcriptase inhibitors (NNRTI) originated by Idenix (acquired by Merck & Co.). It had been in phase II clinical trials for the treatment of HIV infection. However, in 2010, the compound was placed on clinical hold by the FDA. In 2009, the compound was licensed by Idenix to GlaxoSmithKline for the treatment of HIV infection on a worldwide basis.
PATENT
WO2008/042240 A2, 2008, Compound III
compound 66a: racemic form
5-chloro-3-[ methyl 3-((Zζ)-2-cyanovinyl)-5-methylphenyl] phosphinoyl-l//-indole-2- carboxamide.

[00258] Compound 66a was synthesized according to method AL. White solid, 1H NMR (CDCl3, 300 MHz) δ 2.40 (s, 3H), 3.88 (d, J= 11.7 Hz, 3H), 5.89 (d, J= 16.5 Hz, IH), 5.97 (brs, IH), 7.33-7.67 (m, 7H), 10.46 (s, IH), 10.89 (brs, IH), 31P NMR (CDCl3, 121.49 MHz) δ 31.54. MS (ES+) m/z = 414 (MH+).
Example 8

Preparation of Compound HI

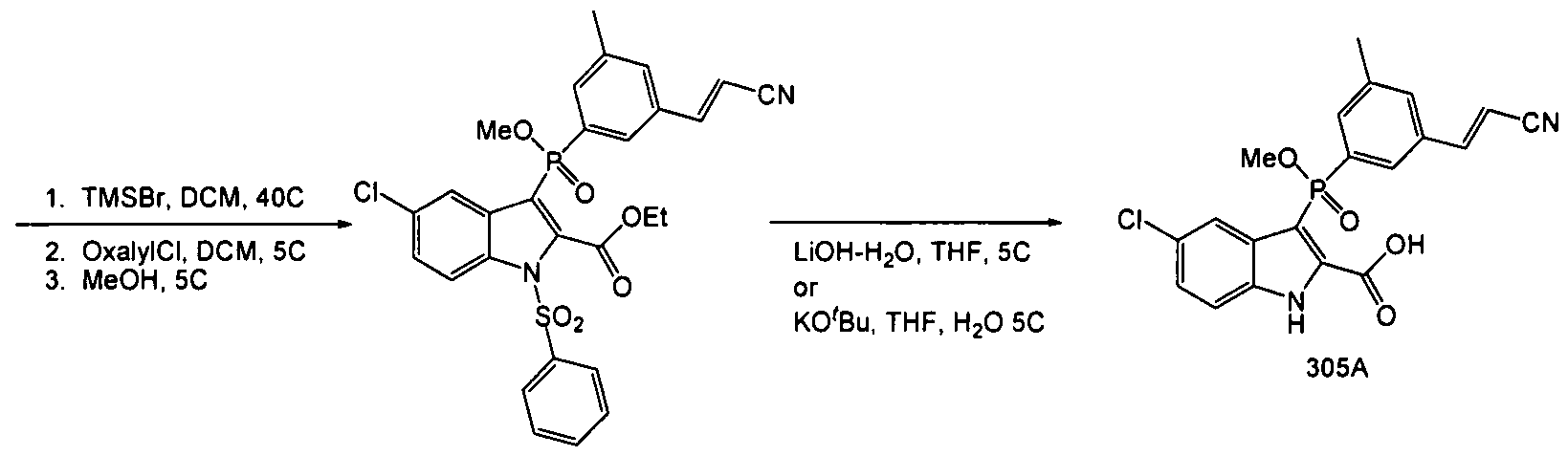
305
1 (-)cιnchonιdιne, Acetone
2 1N HCI1 EtOAc

Compound 302
[00348] A suitable reactor was charged Compound 301 (10Og, 0.23mol) and tetrahydrofuran (IL). The resulting solution was chilled between -90° to -100°C under nitrogen using a LN2 / IPA slush bath, then was treated with n-butyl lithium (2.5M in Hexanes, 99ml, 0.25mol) added over 10 minutes. To this was added diethyl chlorophosphite (37.1g, 0.24mol) over 10 minutes. HPLC (Method 001, RT = 18.9 min) showed no starting material and ca. 85% product. The reaction was then diluted with ethyl acetate (IL) and was allowed to warm to -4O0C. The mix was then treated with hydrochloric acid (0.5M, 590ml) and was allowed to warm to ambient temperature and stir for 30 minutes. The resulting layers were separated and the aqueous extracted with ethyl acetate (500ml). The organics were combined and washed with brine (500ml) dried over sodium sulfate, filtered and concentrated to an oil. 88% HPLC AUC (Method 20, RT = 5.8 min) 115g, >100% yield due to impurities and solvent. Used as is in the next step. Compound 303
[00349] A suitable reactor was charged with Compound 302 (111 g, estimated 0.18mol), iodocinnamonitrile (47.1g, 0.175mol), triethylamine (29.3ml, 0.21mol) and toluene (800ml). The resulting mix was degassed by sparging with a stream of nitrogen for 10 minutes at ambient temperature, after which time tetrakis(triphenylphosphine) palladium(O) (10. Ig, 0.0088mol) was added. The mix was sparged for an additional 5 minutes, then was heated to 80°C for 2 hours. HPLC (Method 20, RT = 6.5 min) showed a complete reaction. The mix was cooled to ambient and was filtered through celite and washed with ethyl acetate (400ml). The combined organics were washed with brine (2 x 500ml) then dried over sodium sulfate, filtered and concentrated to a volume of 350ml. The concentrate was cooled to O0C and was stirred for 1 hour, during which time the product crystallized. The solids were filtered and washed with hexane:toluene (2:1, 150ml). Dried to leave 95g, 90% yield, HPLC AUC 98% (Method 20). Used as is in the next reaction. [00350] 303: C29H26ClN2O6PS 597.02gmol“‘ m/z (ESI+): 597.0 (MH+, 100%), 599.0 (MH+, 35%) 1H NMR δH (400 MHz, CDCl3): 1.38, 1.48 (2 x 3H, 2 x t, COOCH2CH3, POOCH2CH3), 2.41 (3Η, s, Ar-CH3), 4.09-4.16 (2Η, m, POOCH2CH3), 4.52 (2H, q, COOCH2CH3), 5.93 (IH, d, CH=CHCN), 7.33-7.38 (3Η, m, CH=CHCN, 2 x Ar-H), 7.52 (2Η, t, 2 x Ar-H), 7.64 (1Η, t, Ar-H), 7.74, 7.77 (2 x 1Η, 2 x d, 2 x Ar-H), 7.85 (1Η, d, Ar- H), 7.94 (1Η, dd, Ar-H), 8.08 (2Η, d, 2 x Ar-H) 1H NMR δH (400 MHz, d6-DMSO): 1.26, 1.33 (2 x 3H, 2 x t, COOCH2CH3, POOCH2CH3), 2.34 (3Η, s, Ar-CH3), 3.95-4.10 (2Η, m, POOCH2CH3), 4.40 (2H, q, COOCH2CH3), 6.52 (IH, d, CH=CHCN), 7.52 (1Η, dd, Ar-H), 7.60-7.84 (8Η, m, CH=CHCN, 7 x Ar-H), 8.07 (3 x 1Η, m, 3 x Ar-H)
Compound 304
[003511 A suitable reactor was charged with Compound 303 (537g, 0.90mol) and methylene chloride (2.0L). The resulting solution was cooled to O0C, and was treated with bromotrimethylsilane (45Og, 2.9mol) added over 15 minutes. The reaction was then warmed to 400C for 1.5 hours. ΗPLC (Method 20, RT = 4.4 min) indicated a complete reaction. The excess TMSBr was stripped under vacuum (40 – 45°C) and the resulting sticky solid was resuspended in DCM (2.5L) and chilled to 00C. Oxalyl chloride (156ml, 1.8mol) was added over 15 minutes, followed by N,N-dimethylformamide (13.7ml, 0.18mol) both added at O0C. Gas evolution was observed during the DMF addition. After 1 hour, ΗPLC (Method 20, RT = 6.2 min, sample quenched with anhydrous methanol prior to injection) showed a complete reaction. The solvents were stripped again to remove residual oxalyl chloride and the mix resuspended in chilled methanol (3.0L) at 0° – 5°C, and then was allowed to warm to ambient. After two hours, HPLC indicated a complete reaction (HPLC Method 20, RT = 6.2 min). The solution was concentrated to a volume of 1.5L, and the resulting thin slurry was cooled to 0°C, and was diluted with an aqueous solution of sodium bicarbonate (126g, 3L water). After 2 hours at 50C, the product was filtered and washed with cold water / methanol (2:1, 1.5L) then dried to leave 50Og Compound 304. HPLC (Method 20) purity 92% used as is.
Compound 305
[00352] A suitable reactor was charged with Compound 304 (ca. 28Og, 0.48mol) and tetrahydrofuran (2.8L). The resulting solution was then cooled to 5°C and was treated with lithium hydroxide monohydrate (45g, 1.07mol) added in one portion. The reaction was allowed to warm to ambient, during which time the color lightened and a white precipitate formed. After overnight stirring, HPLC indicated an incomplete reaction (Method 20, product RT = 4.3, partially deprotected RT = 5.1, major impurity RT = 3.8). An additional 10% LiOH-H2O was added, but after 10 hours, the partially deprotected intermediate remained at 5%, and the impurity peak at 3.8 minutes had increased to ca. 25%. The reaction was cooled to 50C and was acidified with hydrochloric acid (5N, 280ml) then was diluted with ethyl acetate (2L). The layers were separated and the aqueous extracted with ethyl acetate (500ml). The combined organics were washed with brine (IL) and dried with sodium sulfate, then concentrated to leave a crude oily solid, Compound 305. Ca. 300g, HPLC AUC 57%.
[00353] The crude product was taken up in acetonitrile (1.2L) at 4O0C, and the product triturated w/ water (1.2L). The resulting slurry was cooled to 50C and was allowed to granulate for 30 minutes, after which time the product was filtered and washed with ACN:H2O (1 :1, 100 ml). Ca. 103g, 88% by HPLC. The product was then recrystallized from 360ml ACN at 400C and 360ml water as before. Filtered, washed and dried to leave 75g Compound 305. HPLC AUC 97%. Used as is in the next step.
Compound 306 (chiral resolution)
[00354] A suitable reactor was charged with Compound 305 (28Og, 0.66mol) and acetone (4.2L). The resulting thin slurry was then treated with (-)-cinchonidine (199g, 0.66mol) added in one portion. After one hour, a solution had formed, and after an additional hour, a white solid precipitated, and the mix was left to stir for an additional two hours (four hours total) after which time the solids were filtered, washed with acetone (200ml) and dried to leave 199g Crude Compound 306 cinchonidine salt. HPLC showed an isomer ratio of 96:4.
[00355] The crude salt was then slurried in ethyl acetate (3L) and hydrochloric acid (IN, 3L). The two phase solution was vigorously stirred for 2 hours at ambient temperature. The layers were separated, and the aqueous extracted with ethyl acetate (3L). The organics were combined, dried with sodium sulfate, and concentrated to leave the free base Compound 306, 107g, 95:5 by chiral HPLC.
[00356] The crude Compound 306 was then suspended in acetone (1.07L) and treated with (-)-cinchonidine (76g, 0.26 mol.) After 4 hours total stir time (as above) the solids were filtered, washed with acetone (200ml) and dried to leave 199g of the salt. HPLC 98.6:1.4.
[00357] The salt was broken by dissolving in ethyl acetate (3L) and hydrochloric acid (IN, 3L). The two phase solution was stirred for 2 hours at ambient temperature. The layers were separated, and the aqueous extracted with ethyl acetate (2L). The organics were combined, dried with sodium sulfate, and concentrated to leave the free base Compound 306, 98g, 98.6:1.4 by chiral HPLC. 70% recovery of the desired isomer, 35% yield from the racemic Compound 306. #6: C20H16ClN2O4P 414.78gmol“‘ m/z (ESI+): 415.1 (MH+, 100%), 417.0 (MH+, 35%) [α]D 25 : -47.51 (c, 10.66mgml“‘ in EtOAc) [Opposite enantiomer [α]D 25 : +47.26 (c, 9.60mgml“‘ in EtOAc)] 1H NMR δH (400 MHz, d6-DMSO): 2.33 (3 H, s, Ar-CH3), 3.71 (3H, d, CH3OP), 6.50 (1Η, d, CH=CHCN), 7.36 (1Η, dd, H-6), 7.57 (1Η, d, H-I), 7.66-7.71 (2Η, m, H-4, Ar-Hortho), 7.67 (1Η, d, CH=CHCN), 7.84 (IH, d, Ar-Hortho), 7.98 (1Η, s, Ar-Hpara), 12.97 (1Η, s, N-H), 14.38 (1Η, br-s, COOH) Multiple δc values indicate splitting of carbon signal due to P. 13C NMR δc (100 MHz, d6-DMSO): 20.68 (Ar-CH3), 51.70 (CH3OP), 98.15 (CH=CHCN), 102.33, 103.85, 1 14.98, 120.91 (3 x Q, 118.47 (CN), 125.39 (C), 126.78 (Q, 127.74, 127.86 (C- Hortho), 129.78, 129.88 (Q, 131.25 (Q, 132.06 (Q, 133.44, 133.55 (Q, 133.89, 134.05 (Q, 134.62, 134.75 (Q, 135.47, 135.66 (Q, 138.78, 138.91 (Q, 149.62 (CH=CHCN), 160.40 (C=O) 31P NMR δP (162 MHz, d6-DMSO): 33.50 (IP, s)
Compound HI
[00358] A suitable reactor was charged with Compound 306 (0.63g, O.OOHmol) and 1 ,2-dimethoxyethane (10ml.) The mix was treated with 1,1-carbonyldiimidazole (0.47g, 0.0028mol) added in one portion, and the mix was allowed to stir at ambient temperature until gas evolution ceased (ca. 1.5 hours.) The solution was then cooled to 50C, and was sparged with ammonia gas for 5 minutes. HPLC (Method 20, product RT=5.0 min) showed a complete reaction after one hour at ambient. The reaction was quenched by the addition of 1Og crushed ice, and was concentrated under reduced pressure to remove the DME. The resulting slurry was stirred for one hour at 50C to granulate the product. The solids were filtered and dried to leave pure Compound III ((2-Carbamoyl-5-chloro-4-fluoro-lH-indol-3- yl)-[3-((E)-2-cyano-vinyl)-5-methyl-phenyl]-(S)-phosphinic acid methyl ester) as a white solid 0.56g, 89% yield. HPLC (Method 20) chemical purity 98.5%. Chiral purity 97%. [00359] A suitable reactor was charged with Compound 306 (1Og, 0.024mol) and 1,2- dimethoxyethane (150ml). The mix was treated with 1,1-carbonyldiimidazole (7.8g, 0.048mol) added in one portion, and the mix was allowed to stir at ambient temperature until gas evolution ceased. The solution was then cooled to 5°C, and was sparged with ammonia gas for 5 minutes. HPLC (Method 20, product RT=5.0 min) showed a complete reaction after one hour. The reaction was quenched by the addition of lOOg crushed ice, and was concentrated under reduced pressure to remove the DME. The resulting oily solid (in water) was diluted with methanol (20ml) and stirred for one hour at 50C to granulate the product. The solids were filtered and dried to leave pure Compound III ((2-Carbamoyl-5- chloro-4-fluoro-lH-indol-3-yl)-[3-((E)-2-cyano-vinyl)-5-methyl-phenyI]-(S)-phosphinic acid methyl ester). 9.8g, 98% yield. HPLC (Method 20) chemical purity 99.5%. Chiral purity 94.3%.
Compound III: C20Hi7ClN3O3P 413.79gmol“‘ m/z (ESI+): 414.1 (MH+, 100%), 416.1 (MH+, 35%)
vmax (KBr disc) (cm“1) 1620.0 (amide I), 1670.6 (amide II), 2218.7 (CN), 3125.5, 3291.9 (N-H)
[α]D 20 : -75.08 (c, 9.04mgmr’ in CHCl3)
m.p.: 144- 1480C transition to opaque semi-solid, 209-2100C melts
Elemental analysis: C20H17ClN3O3P calculated C 58.05%, H 4.14%, N 10.15%, Cl 8.57%, P 7.49%. Found C 58.13%, H 4.08%, N 10.16%, Cl 8.69%, P 7.44%
1H NMR δH (400 MHz, d6-DMSO): 2.32 (3H, s, Ar-CH3), 3.74 (3Η, d, CH3OP), 6.52 (1Η, d, CH=CHCN), 7.30 (1Η, dd, H-6), 7.53-7.58 (3Η, m, H-4, H-7, H-6′), 7.68 (1Η, d, CH=CHCN), 7.73 (IH, s, H-4′), 7.75 (1Η, d, H-2′), 8.02, 10.15 (2 x 1Η, 2 x s, NH2), 12.80 (1Η, s, N-H) Multiple δc values indicate splitting of carbon signal due to P.
13C NMR δc(100 MHz, d6-DMSO): 20.77 (Ar-CH3), 51.75, 51.81 (CH3OP), 98.39, 98.91 (C-3), 98.44 (CH=CHCN), 1 15.05 (C-7), 1 18.53 (CN), 119.96 (C-4), 124.73 (C-6), 126.68 (C-5), 127.15, 127.26 (C-2′), 129.25, 129.35 (C-9), 131.37 (C-4′), 132.45, 134.04 (C-I ‘), 132.69, 132.80 (C-6′), 133.92 (C-8), 134.30, 134.44 (C-3′), 139.33, 139.46 (C-5’), 139.96, 140.17 (C-2), 149.55 (CH=CHCN), 160.65 (C=O)
31P NMR δP (162 MHz, d6-DMSO): 33.72 (IP, s)
PATENT
http://www.google.ch/patents/WO2009120914A1?cl=en&hl=de

(2-carbamoyl-5-chloro-lH-indol-3-yl)-[3-((E)-2-cyano-vinyl)-5-methyl-phenyl]- (7?)-phosphinic acid methyl ester (I):
| WO2008042240A2 * | 28. Sept. 2007 | 10. Apr. 2008 | Idenix Pharmaceuticals, Inc. | Enantiomerically pure phosphoindoles as hiv inhibitors |
| US20060074054 * | 16. Sept. 2005 | 6. Apr. 2006 | Richard Storer | Phospho-indoles as HIV inhibitors |
Figure 7 provides an infrared spectrum of Form I.
Paper
Development of an Efficient Manufacturing Process to GSK2248761A API
 , John D. Hayler† , Alan Ironmonger†, David Pascoe†, Neil Hodnett†, Jonathan Stanway†, Stephen Etridge†, Alistair Stewart‡⊥, Jingyang Wang‡⊥, Benjamin A. Mayes‡⊥, and Adel Moussa‡⊥
, John D. Hayler† , Alan Ironmonger†, David Pascoe†, Neil Hodnett†, Jonathan Stanway†, Stephen Etridge†, Alistair Stewart‡⊥, Jingyang Wang‡⊥, Benjamin A. Mayes‡⊥, and Adel Moussa‡⊥

Amidation of indole 2-carboxylate 1 with ammonia gas via the imidazolide 2 gave GSK2248761A API 3, which was in development for the treatment of HIV. Three significant impurities, namely the phosphinic acid 4, the N-acyl urea 8, and the indoloyl carboxamide 6, were formed during the reaction, and the original process was unable to produce API within clinical specification when run at scale. Investigation into the origin, fate, and control of these impurities led to a new process which was able to produce API within clinical specification.
Development and Scale-Up of a Manufacturing Route for the Non-nucleoside Reverse Transcriptase Inhibitor GSK2248761A (IDX-899): Synthesis of an Advanced Key Chiral Intermediate
, John Hayler† , Neil Hodnett†, Alan Ironmonger*†, Augustine Ochen†, David Pascoe†, John Richardson†§, Erica Vit†§, François-René Alexandre‡ , Catherine Caillet‡∥, Agnès Amador‡∥, Stéphanie Bot‡, Séverine Bonaric‡∥, Daniel da Costa‡, Marie-Pierre Lioure‡, Arlène Roland‡∥, Elodie Rosinovsky‡∥, Christophe Parsy‡∥, and Cyril B. Dousson‡Abstract

A new and improved synthetic route to an intermediate in the synthesis of the phosphinate ester GSK2248761A is described. In the key step, we describe the first process-scale example of a palladium-catalyzed phosphorus–carbon coupling to give the entire backbone of GSK2248761A in one telescoped stage in 65% average yield on a 68 kg scale. This unusual chemistry enabled the route to be reduced from six chemistry stages to four and eliminated a number of environmentally unfriendly reagents and solvents.
REFERENCES
1: Dousson C, Alexandre FR, Amador A, Bonaric S, Bot S, Caillet C, Convard T, da Costa D, Lioure MP, Roland A, Rosinovsky E, Maldonado S, Parsy C, Trochet C, Storer R, Stewart A, Wang J, Mayes BA, Musiu C, Poddesu B, Vargiu L, Liuzzi M, Moussa A, Jakubik J, Hubbard L, Seifer M, Standring D. Discovery of the Aryl-phospho-indole IDX899, a Highly Potent Anti-HIV Non-nucleoside Reverse Transcriptase Inhibitor. J Med Chem. 2016 Feb 3. [Epub ahead of print] PubMed PMID: 26804933.
2: Margolis DA, Eron JJ, DeJesus E, White S, Wannamaker P, Stancil B, Johnson M. Unexpected finding of delayed-onset seizures in HIV-positive, treatment-experienced subjects in the Phase IIb evaluation of fosdevirine (GSK2248761). Antivir Ther. 2014;19(1):69-78. doi: 10.3851/IMP2689. Epub 2013 Oct 24. PubMed PMID: 24158593.
3: Ölgen S. Recent development of new substituted indole and azaindole derivatives as anti-HIV agents. Mini Rev Med Chem. 2013 Oct;13(12):1700-8. Review. PubMed PMID: 23895189.
4: Castellino S, Groseclose MR, Sigafoos J, Wagner D, de Serres M, Polli JW, Romach E, Myer J, Hamilton B. Central nervous system disposition and metabolism of Fosdevirine (GSK2248761), a non-nucleoside reverse transcriptase inhibitor: an LC-MS and Matrix-assisted laser desorption/ionization imaging MS investigation into central nervous system toxicity. Chem Res Toxicol. 2013 Feb 18;26(2):241-51. doi: 10.1021/tx3004196. Epub 2012 Dec 20. PubMed PMID: 23227887.
5: Zala C, St Clair M, Dudas K, Kim J, Lou Y, White S, Piscitelli S, Dumont E, Pietropaolo K, Zhou XJ, Mayers D. Safety and efficacy of GSK2248761, a next-generation nonnucleoside reverse transcriptase inhibitor, in treatment-naive HIV-1-infected subjects. Antimicrob Agents Chemother. 2012 May;56(5):2570-5. doi: 10.1128/AAC.05597-11. Epub 2012 Feb 6. PubMed PMID: 22314532; PubMed Central PMCID: PMC3346662.
6: Piscitelli S, Kim J, Gould E, Lou Y, White S, de Serres M, Johnson M, Zhou XJ, Pietropaolo K, Mayers D. Drug interaction profile for GSK2248761, a next generation non-nucleoside reverse transcriptase inhibitor. Br J Clin Pharmacol. 2012 Aug;74(2):336-45. doi: 10.1111/j.1365-2125.2012.04194.x. PubMed PMID: 22288567; PubMed Central PMCID: PMC3630753.
7: La Regina G, Coluccia A, Silvestri R. Looking for an active conformation of the future HIV type-1 non-nucleoside reverse transcriptase inhibitors. Antivir Chem Chemother. 2010 Aug 11;20(6):213-37. doi: 10.3851/IMP1607. Review. PubMed PMID: 20710063.
8: Klibanov OM, Kaczor RL. IDX-899, an aryl phosphinate-indole non-nucleoside reverse transcriptase inhibitor for the potential treatment of HIV infection. Curr Opin Investig Drugs. 2010 Feb;11(2):237-45. Review. PubMed PMID: 20112173.
9: Zhou XJ, Garner RC, Nicholson S, Kissling CJ, Mayers D. Microdose pharmacokinetics of IDX899 and IDX989, candidate HIV-1 non-nucleoside reverse transcriptase inhibitors, following oral and intravenous administration in healthy male subjects. J Clin Pharmacol. 2009 Dec;49(12):1408-16. doi: 10.1177/0091270009343698. Epub 2009 Sep 23. PubMed PMID: 19776293.
10: Zhou XJ, Pietropaolo K, Damphousse D, Belanger B, Chen J, Sullivan-Bólyai J, Mayers D. Single-dose escalation and multiple-dose safety, tolerability, and pharmacokinetics of IDX899, a candidate human immunodeficiency virus type 1 nonnucleoside reverse transcriptase inhibitor, in healthy subjects. Antimicrob Agents Chemother. 2009 May;53(5):1739-46. doi: 10.1128/AAC.01479-08. Epub 2009 Feb 17. PubMed PMID: 19223643; PubMed Central PMCID: PMC2681571.
11: Mascolini M, Larder BA, Boucher CA, Richman DD, Mellors JW. Broad advances in understanding HIV resistance to antiretrovirals: report on the XVII International HIV Drug Resistance Workshop. Antivir Ther. 2008;13(8):1097-113. PubMed PMID: 19195337.
12: Dalton P. Two new NNRTIs enter the pipeline. Proj Inf Perspect. 2008 Sep;(46):13. PubMed PMID: 19048672.
13: Sweeney ZK, Klumpp K. Improving non-nucleoside reverse transcriptase inhibitors for first-line treatment of HIV infection: the development pipeline and recent clinical data. Curr Opin Drug Discov Devel. 2008 Jul;11(4):458-70. Review. PubMed PMID: 18600563.
/////////////GSK2248761A , IDX899, Fosdevirine, PHASE 2
CC1=CC(=CC(=C1)C=CC#N)P(=O)(C2=C(NC3=C2C=C(C=C3)Cl)C(=O)N)OC