














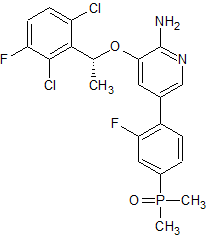
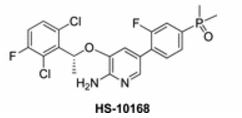
HS-10168
CAS 1394346-15-6
471.26, C21 H19 Cl2 F2 N2 O2 P
(R)-(4-(6-Amino-5-(1-(2,6-dichloro-3-fluorophenyl)ethoxy)pyridin-3-yl)-3-fluorophenyl)dimethylphosphine Oxide (HS-10168)
2-Pyridinamine, 3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-[4-(dimethylphosphinyl)-2-fluorophenyl]-
HS-10168 is a potent and selective mesenchymal epithelial transition factor/anaplastic lymphoma kinase (c-Met/ALK) inhibitor that has advanced into Phase I clinical trials.(1, 2)
| Inventors | Baihua Hu, Kan He, Minsheng Zhang |
| Applicant | Jiangsu Hansoh Pharmaceutical Co., Ltd. |

Kinases are a superfamily of enzymes that transfer a phosphate group from ATP to target proteins. There are more than 518 kinases encoded in the human genome, including 90 tyrosine kinases, 388 serine/threnine kinases and 40 atypical kinases (Manning, G., D. B. Whyte, et al. (2002), “The protein kinase complement of the human genome” Science 298(5600): 1912-1934). They play vital roles in cell activation, proliferation, differentiation, migration, vascular permeability, etc. Dysfunction of kinases has been implicated in various diseases such as cancer, inflammation, cardiovascular diseases, diabetes, and neuronal disorders. Several kinase inhibitors have been developed for the treatment of cancers, including but not limited to imatinib, dasatinib, nilotinib, gefitinib, erlotinib, lapatinib, sunitinib, sorafenib, pazopanib, evrolimus, trastuzumab, cetuximab, panitumumab,
bevacizumab (Knight, Z. A., H. Lin, et al. (2010). “Targeting the cancer kinome through polypharmacology” Nat. Rev. Cancer 10(2): 130-137).
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase in the insulin receptor family. Other members of the family include lymphocyte tyrosine kinase, insulin receptor kinase, IGF-1 receptor kinase, RTK neutrophin receptor kinases and hepatocyte growth factor/scatter factor (Met) kinase. ALK, which was initially discovered by cloning of the nucleolar protein nucleophosmin (NPM)-ALK fusion gene in anaplastic large cell lymphomas, is encoded by a genomic locus at the chromosomal band 2p23 in the human (Morris, S. W., M. N. Kirstein, et al.
(1994). “Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non- Hodgkin’s lymphoma” Science 263(5151 ): 1281 -1284; Shiota, M., J. Fujimoto, et al. (1994). “Hyperphosphorylation of a novel 80 kDa protein-tyrosine kinase similar to Ltk in a human Ki-1 lymphoma cell line, AMS3” Oncogene 9(6): 1567-1574). The genes encoding native, full length receptor forms of ALK in human and mouse were cloned in 1997 (Iwahara, T., J. Fujimoto, et al. (1997). “Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system” Oncogene 14(4): 439-449; Morris, S. W., C. Naeve, et al. (1997). “ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK)” Oncogene 14(18): 2175-2188). The single chain of native ALK protein consists of 1620 amino acids with posttranslational modifications, forming LDL-A, MAM, glycine-rich, transmembrane, and catalytic domains. There are three tyrosine residues (Tyr1278, Tyr1282 and Tyr1283) forming the autophosphorylation motif YxxxYY in the activation loop, a common structural feature found with insulin and IGF1 receptor kinases. The sequential phosphorylation of this tyrosine triplet regulates kinase activity. Recently, X-ray crystal structures of the ALK catalytic domain were determined in apo, ADP-, or inhibitor-bound forms (Bossi, R. T., M. B. Saccardo, et al. (2010). “Crystal structures of anaplastic lymphoma kinase in complex with ATP competitive inhibitors.” Biochemistry 49(32): 6813-6825; Lee, C. C, Y. Jia, et al. (2010). “Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain” Biochem J 430(3): 425-437; Mctigue, M., Y. Deng, et al. (2010). ” Structure of the human anaplastic lymphoma kinase in complex with crizotinib (PF- 02341066)” Protein database (2XP2)). ALK shares the basic tyrosine kinase domain architecture and topology. A small N-terminal lobe is connected to a larger C- terminal lobe by a loop referred to as the hinge region, in which E1 197 and M 1 199 forms important hydrogen bonds with ATP/ADP and inhibitors. The activation loop, which consists of residues 1270-1299, begins with the DFG-motif and ends with residues PPE. The catalytic loop, which consist of residues 1247-1254, positions between alphaE and the first strand of the 2-stranded beta-sheet. The structures reveal important interactions between active site residues and inhibitors, and how the functional mutants affect the kinase activity.
The native ALK is dominantly expressed in the central and peripheral nervous systems during development (Iwahara, T., J. Fujimoto, et al. (1997).
“Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system” Oncogene 14(4): 439-449; Morris, S. W., C. Naeve, et al. (1997). “ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK)” Oncogene 14(18): 2175-2188). As reported by Iwahara et al., the ALK mRNA was detected in thalamus, hypothalamus, mid brain, dorsal root ganglia and olfactory bulb in mouse from day 1 1 . However, the expression level decreased near the gestation, and become barely detectable in adult mouse. ALK expression was only observed in rare scattered neural cells, endothelial cell and pericytes in nervous system in adult and human tissues
(Iwahara, T., J. Fujimoto, et al. (1997). “Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system” Oncogene 14(4): 439- 449; Pulford, K., L. Lamant, et al. (1997). “Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1 ” Blood 89(4): 1394-1404). The restricted tissue expression pattern suggests that ALK plays an important role in the development and function of nervous system. Consistently, ALK receptor was demonstrated as the receptor for growth factors pleiotrophin and midkine for neurite outgrowth (Stoica, G. E., A. Kuo, et al. (2001 ). “Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin” J. Biol. Chem. 276(20):
16772-16779; Stoica, G. E., A. Kuo, et al. (2002). “Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types” J. Biol. Chem. 277(39): 35990-35998; Yanagisawa, H., Y. Komuta, et al. (2010).
“Pleiotrophin induces neurite outgrowth and up-regulates growth-associated protein (GAP)-43 mRNA through the ALK/GSK3beta/beta-catenin signaling in developing mouse neurons” Neurosci. Res. 66(1 ): 1 1 1 -1 16). Furthermore, the ALK knockout mice displayed an increased struggling time in the tail suspension test and the Porsolt swim test and enhanced performance in a novel object-recognition test (Bilsland, J. G., A. Wheeldon, et al. (2008). “Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications” Neuropsychopharmacology 33(3): 685-700). An age-dependent increase in basal hippocampal progenitor proliferation was observed, similar to what is observed after chronic treatment with antidepressants. Other than that, the animals developed normally with no anatomical abnormalities and a full life span. Collectively, these results suggest that ALK could be a new therapeutic target for psychiatric indications, such as schizophrenia and depression.
Overexpression, mutation and fusion proteins of ALK have been implicated in several cancers, including but not limited to neuroblastoma, anaplastic large-cell lymphoma (ALCL), non-small cell lung cancer (NSCLC) and inflammatory myofibroblastic tumor (IMT). When its kinase activity is constitutively enhanced by point mutation, amplification or rearrangement of the corresponding genes, ALK become an oncogenic driver, activating numerous signaling pathways to promote tumorigenesis (Palmer, R. H., E. Vernersson, et al. (2009). “Anaplastic lymphoma kinase: signalling in development and disease” Biochem. J. 420(3): 345-361 ). The signal pathways include those involving Ras and mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), protein kinase B (Akt), and target of rapamycin (TOR), sonic hedgehog (Shh), phospholipase Cy (PLCy), JUN kinase, Janus kinase (JAK) and signal transducer and activator of transcription (STAT).
Neuroblastoma is an embryonal tumor of the peripheral sympathetic nervous system, accounting for approximately 15% of all deaths due to childhood cancer. Overexpression and point mutations of full-length ALK plays an important role in the pathogenesis of neuroblastoma (Chen, Y., J. Takita, et al. (2008).
Oncogenic mutations of ALK kinase in neuroblastoma” Nature 455(7215): 971 -974; George, R. E., T. Sanda, et al. (2008). “Activating mutations in ALK provide a therapeutic target in neuroblastoma” Nature 455(7215): 975-978; Janoueix-Lerosey, Lequin et al. 2008. “Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma” Nature 455(7215): 967-970; Mosse, Y. P., M.
Laudenslager, et al. (2008). “Identification of ALK as a major familial neuroblastoma predisposition gene” Nature 455(7215): 930-935; Passoni, L., L. Longo, et al. (2009). “Mutation-independent anaplastic lymphoma kinase overexpression in poor prognosis neuroblastoma patients.” Cancer Res. 69(18): 7338-7346). There are copy number increases of ALK in more than 25% and mis-sense mutations in 6-8% of the primary neuroblastomas. The mutants identified include but not limited to F1 174L, R1257Q, G1 128A, M 1 166R, 11 171 N, F1 174I, R1 192P, F1245C, F1245V, I1250T, 11250V, T1087I, K1062M and Y1278S. Among of them, F1 174L is the most frequent somatic mutant, identified in approximately 4% of primary tumors. R1257Q is the mutant identified in both familial and sporadic tumors. Most of the mutations map to critical regions of the kinase domain and are oncogenic drivers. Mutated ALK proteins are over-expressed, hyper-phosphorylated and show constitutive kinase activity in primary neuroblastomas. The knockdown of ALK expression or inhibition of ALK activity by small molecule inhibitors in ALK-mutated cells, and in cell lines over-expressing a wild-type ALK, led to a marked decrease of cell proliferation. Altogether, the available data identify ALK as a critical player in neuroblastoma development, and may represent a very attractive therapeutic target for the treatment of this disease that is still frequently fatal with current treatments.
ALCL is a rare form of indolent Non-Hodgkin’s lymphoma that affects T- cells. It is more common in children and men. ALCL often affects the lymph nodes, skin, liver, lungs, and bone marrow. This disease can be either systematic or cutaneous. Approximately 60-80% ACLC is ALK positive (Morris, S. W., M. N.
Kirstein, et al. (1994). “Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma” Science 263(5151 ): 1281 -1284). The most frequent ALK fusion protein is NPM-ALK, being found in 75-80% of all ALK-positive ALCL patients. Other ALK fusion proteins include but not limited to TPM3-, ATIC-, CLTC-, TFGL-, TFG-, TMP4-ALK. The CLTC-, NPM- or TMP3-ALK (Webb, T. R., J. Slavish, et al. (2009). “Anaplastic lymphoma kinase: role in cancer pathogenesis and small-molecule inhibitor development for therapy” Expert Rev Anticancer Ther. 9(3): 331 -356). ALK-fusion proteins are also found in rare cases of diffuse large B-cell lymphoma and systemic histiocytosis. ALK fusion proteins mediate oncogenesis by activating the classical receptor tyrosine kinase pathway, and most relevantly, the STAT3 phosphorylation and activation. Transgenic mouse expressing NPM-ALK develop large cell lymphoma with a T-cell phenotype and frequent expression of CD30 antigen. ALK has been shown to be a valid therapeutic target for ALCL.
Lung cancer is the leading cause of cancer mortality in the world today. Approximately 85% of the lung cancer is non-small cell lung cancer (NSCLC). ALK gene rearrangement was identified in a small subset (6-7%) of NSCLC patients, involving a small inversion within chromosome 2p to form a fusion gene comprising portions of the echinoderm microtubule-associated protein-like 4 (EML4) gene and the ALK gene (Rikova, K., A. Guo, et al. (2007). “Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer” Cell 131 (6): 1 190-1203; Soda, M., Y. L. Choi, et al. (2007). “Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer” Nature 448(7153): 561 -566). In lung cancer, ALK fusion proteins appear to be restricted to patients with adenocarcinoma, mostly in patients with minimal or no smoking history. ALK abnormalities seem to be mutually exclusive to EGFR and KRAS mutations. Other fusion proteins in NSCLC include but not limited to TGF-ALK, KIF5B-ALK. Auto-phosphorylation of EML4-ALK activates PI3K-AKT and RAS-MAPK pathways, leading to cell growth, proliferation, survival, and cell cycle progression. The oncogenic potential of EML4-ALK was confirmed in transgenic mice that developed hundreds of adenocarcinoma nodules in both lung, and the tumor burden was effectively reduced by administration of an potent ALK inhibitor (Soda, M., S. Takada, et al. (2008). “A mouse model for EML4- ALK-positive lung cancer” Proc. Natl. Acad. Sci. U. S. A. 105(50): 19893-19897). Furthermore, the inhibition of ALK in lung cancer by oral administration of crizotinib, an ALK/cMet inhibitor, resulted in tumor shrinkage or stable disease in most patients. Altogether, ALK is an attractive therapeutic target for NSCLC (Kwak, Bang et al. 2010. “Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer” N. Engl. J. Med. 363(18): 1693-1703).
IMT are uncommon lesions composed of spindled myofibroblasts within a variable background of collagen and inflammatory cells. The frequency of ALK expression in IMT ranges from 36-62%. Several ALK fusion proteins were identified in IMT patients, such as TPM3-, TPM4-, CLTC-, ATIC-, CARS-, RANBP2- and SEC31 L1 -ALK (Webb, T. R., J. Slavish, et al. (2009). “Anaplastic lymphoma kinase: role in cancer pathogenesis and small-molecule inhibitor development for therapy” Expert Rev. Anticancer Ther. 9(3): 331 -356). Other tumors with ALK gene rearrangement include, but not limited to, B-cell Non-Hodgkin Lymphoma, oesophageal squamous cell carcinoma and systemic histiocytosis (Webb, T. R., J. Slavish, et al. (2009). “Anaplastic lymphoma kinase: role in cancer pathogenesis and small-molecule inhibitor development for therapy” Expert Rev. Anticancer Ther. 9(3): 331 -356). With more specific and sensitive assays for ALK detection, it is expected that ALK may play a role in more tumors than those identified so far.
Several small molecule inhibitors of ALK have been reported elsewhere, for example, crizotinib (PF-02341066) is currently under clinical development for lung cancer, ALCL and IMT (Ardini, E., P. Magnaghi, et al. (2010). “Anaplastic Lymphoma Kinase: role in specific tumours, and development of small molecule inhibitors for cancer therapy” Cancer Lett. 299(2): 81 -94; Milkiewicz, K. L. and G. R. Ott (2010). “Inhibitors of anaplastic lymphoma kinase: a patent review.” Expert Opin. Ther. Pat. 20(12): 1653-1681 ). Crizotinib inhibits ALK and cMet activities and proliferation of several ALK positive cancer cell lines. Crizotinib is effective in xenograft cancer models. It is noteworthy that Crizotinib was reported to be a time-dependent cytochrome P450 3A4 inhibitor, causing clinical drug-drug interactions. In the reported phase I study, crizotinib was effective against advanced non-small cell lung cancers carrying activated ALK. The overall response rate was 57% and the rate of stable disease was 33%. The response rate is impressive, as compared with the approximately 10% response rate in such cancers that were treated with second-line chemotherapy. However, two secondary drug-resistance mutations in the catalytic domain, L1 196M and C1 156Y, were observed in a patient who has an initial strong clinical response to crizotinib (Choi, Y. L, M. Soda, et al. (2010). “EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors” N. Engl. J. Med. 363(18): 1734-1739). Each mutation developed independently in sub-clones of the tumor and conferred marked resistance to two different ALK inhibitors. The appearance of crizotinib-resistance mutations indicates that additional ALK inhibitors will be required to target EML4-ALK mutants that are insensitive to crizotinib in a clinical settings (Hallberg, B. and R. H. Palmer (2010). “Crizotinib-latest champion in the cancer wars?” N. Engl. J. Med. 363(18): 1760-1762). [0012] cMet is a high-affinity hepatocyte growth factor receptor (HGF).
cMet/HGF/SF signaling is essential for normal cell proliferation, migration, angiogenesis, embryogenesis, organogenesis, and tissue regeneration
(Danilkovitch-Miagkova, A. and B. Zbar (2002). “Dysregulation of Met receptor tyrosine kinase activity in invasive tumors” J. Clin. Invest. 109(7): 863-867;
Christensen, J. G., J. Burrows, et al. (2005). “c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention” Cancer Lett. 225(1 ): 1 -26). Aberrant cMet/HGF/SF signaling, resulting from mutation or over-expression of the c-Met proto-oncogene and HGF, plays a major role in tumorigenesis, invasion, and metastasis many human tumors. cMet is highly expressed in numerous cancers, and the expression correlates with poor patient prognosis. cMet activating point mutations in the kinase domain are implicated as the cause of hereditary papillary renal carcinoma and were also detected in sporadic papillary renal carcinoma, lung cancers, head and neck cancers, childhood hepatocellular carcinoma, and gastric cancer. Furthermore, amplification of the cMet gene locus was detected in patients with gastric, metastatic colorectal cancer, and esophageal adenocarcinoma. cMet is an attractive therapeutic target for cancer treatment (Christensen, J. G., J. Burrows, et al. (2005). “c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention” Cancer Lett. 225(1 ): 1 -26).
Accordingly, the identification of small-molecules that specifically modulate kinase activity, particularly ALK and/or cMet kinase, serves therapeutic approaches for treatment of cancers, inflammation, cardiovascular and metabolic diseases, psychological and neurological disorders.
SPECTRAL DATA
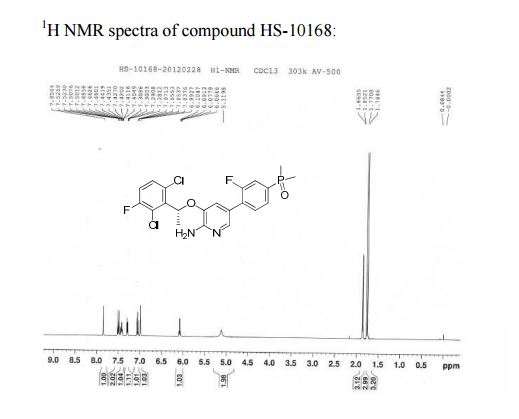
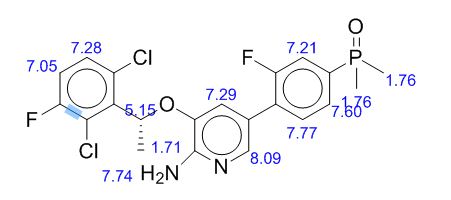
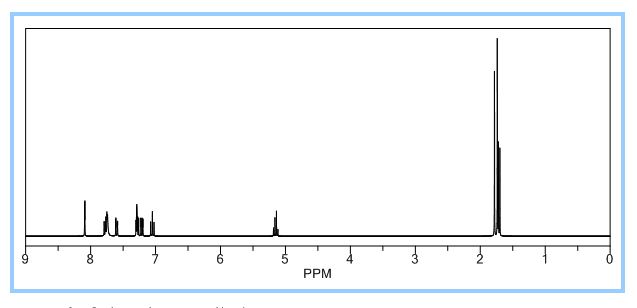
1H NMR (500 MHz, CDCl3) δ (ppm) 1.75 (s, 3H), 1.77 (s, 3H), 1.86 (d, J = 6.7 Hz, 3H), 5.12 (brs, 2H), 6.08 (q, J = 6.6 Hz, 1H), 6.99 (s, 1H), 7.04–7.07 (m, 1H), 7.28–7.31 (m, 1H), 7.40–7.43 (m, 1H), 7.44–7.53 (m, 2H), 7.85 (s, 1H).
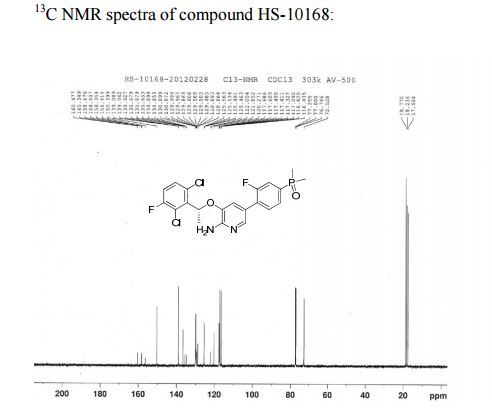
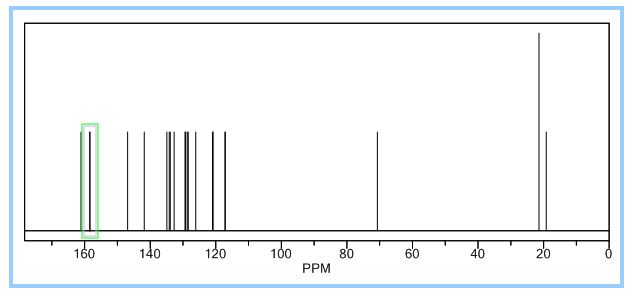
13C NMR (125 MHz): δ (ppm) 17.7, 18.2, 18.8, 72.5, 116.4–116.6, 117.3, 117.4–117.7, 120.3, 121.9–122.0, 125.5–125.6, 128.9, 129.5–129.6, 129.9, 130.0–130.1, 134.9–136.7, 135.6–135.7, 136.7, 139.1, 150.4, 156.4–158.4, 158.3–160.5;
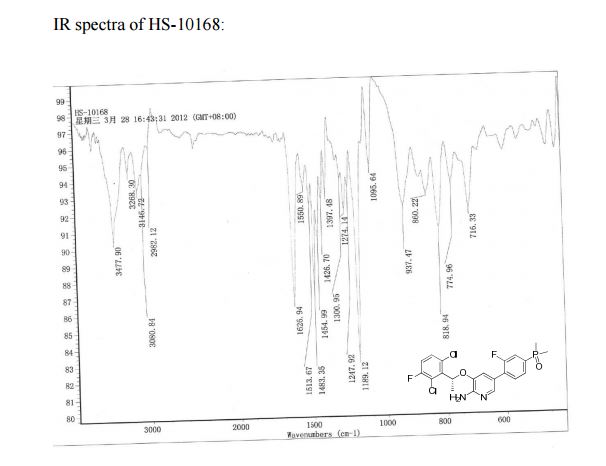
IR (KBr): 3478, 3268, 3081, 2982, 1627, 1514, 1483, 1551, 1455, 1427, 1397, 1189, 1274, 1096, 860, 819, 775 cm–1;
MS(m/z): 471.06 [M + H]+.
syn
Something in colour for students
1H NMR PREDICT
13c nmr predict
Patent
https://www.google.com/patents/WO2012116050A2?cl=en
Example 25
3-[(1 R)-1 -(2,6-dichloro-3-fluoro-phenyl)ethoxy]-5-(4-dimethylphosphoryl-2- fluoro- hen l ridin-2-amine

[0157] The title compound was prepared from 5-bromo-3-[(1 R)-1 -(2,6-dichloro-3- fluoro-phenyl)-ethoxy]-pyridin-2-ylamine, 4-bromo-2-fluoro-phenylboronic acid, and dimethylphosphine oxide followed the same procedures as Example 1 Step 1 and Step 3; ESMS: m/z 471 (M+H)+; chiral purity 93.12% (column AD-H 4.6*250 mm 5 urn; solvent:hexane/isopropanol).
PATENT
https://www.google.com/patents/WO2016019909A1?cl=fi
PAper

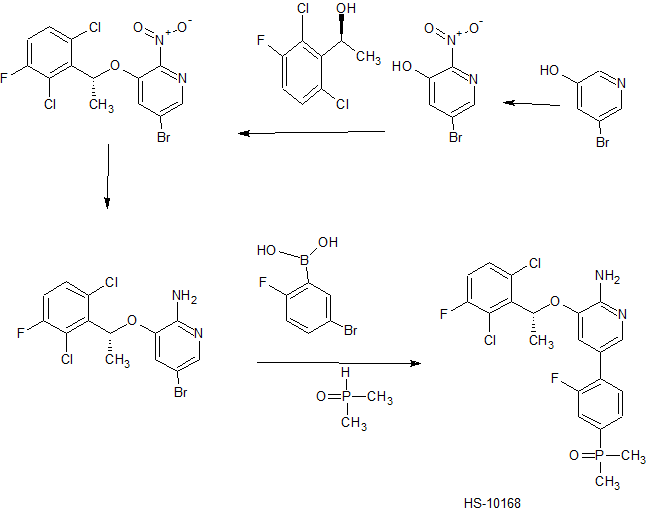
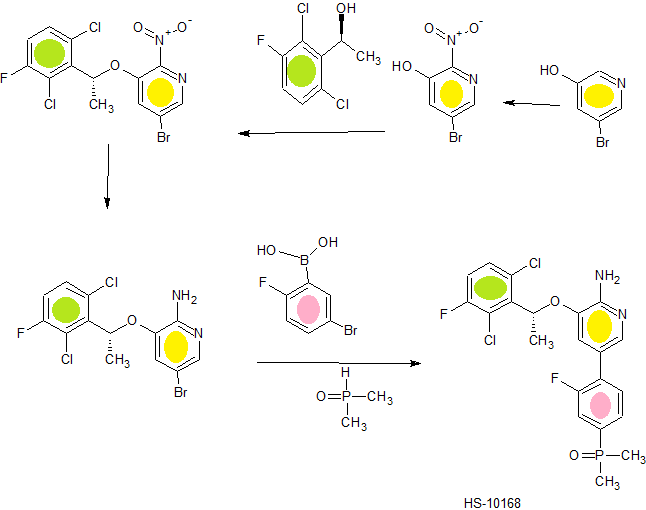
A modified synthetic route to c-Met/ALK inhibitor HS-10168 has been developed on a kilogram scale. The key steps of the new process include a Suzuki coupling reaction of nitro-containing bromopyridine 6 and para-phenol boronate 9 to give intermediate 10, which was then converted to its triflate 11. The phosphorus group was introduced by coupling of 11 and dimethylphoshine oxide 8 to give compound 12, which was reduced to produce HS-10168
Development of a Modified Process for the Kilogram-Scale Synthesis of c-Met/ALK Inhibitor HS-10168
 and JinJia Wang
and JinJia Wang-
(a) de Koning, P. D.; McAndrew, D.; Moore, R.; Moses, I. B.; Boyles, D. C.; Kissick, K.; Stanchina, C. L.;Cuthbertson, T.; Kamatani, A.; Rahman, L.; Rodriguez, R.; Urbina, A.; Sandoval, A.; Rose, P. R. Org. Process Res. Dev. 2011, 15, 1018– 1026, DOI: 10.1021/op200131n
(b) Cui, J. J.;Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.;McTigue, M.; Grodsky, N.; Ryan, K.; Padrique, E.; Alton, G.; Timofeevski, S.; Yamazaki, S.; Yamazaki, S.;Li, Q.; Zou, H.; Christensen, J.; Mroczkowski, B.; Bender, S.; Kania, R. S.; Edwards, M. J. Med. Chem.2011, 54, 6342– 6363, DOI: 10.1021/jm2007613
-
2
Hu, B.; He, K.; Zhang, M. WO2012/116050, 2012.
PATENT
CN 105330697, https://www.google.com/patents/CN105330697A?cl=zh
WO 2016019909
WO 2016019910
///////////HS-10168, HS 10168, PHASE 1
Nc2ncc(cc2O[C@H](C)c1c(Cl)ccc(F)c1Cl)c3ccc(cc3F)P(C)(C)=O