














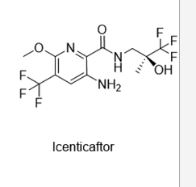
QBW 251, ICENTICAFTOR
- Molecular FormulaC12H13F6N3O3
- Average mass361.240 Da
Icenticaftor (development code QBW251) is a drug candidate for the treatment of chronic obstructive pulmonary disease (COPD)[1][2] and cystic fibrosis.[3][4] The drug is being developed by Novartis.[5]
Like ivacaftor (which is marketed as Kalydeco), icenticaftor functions by acting as a stimulator of the protein cystic fibrosis transmembrane conductance regulator (CFTR).[5]
Icenticaftor (QBW251) is an orally active CFTR channel potentiator, with EC50s of 79 nM and 497 nM for F508del and G551D CFTR, respectively. Icenticaftor can be used for chronic obstructive pulmonary disease (COPD) and cystic fibrosis research.
Cystic fibrosis (CF) is the most prevalent life-threatening Mendelian disorder in Caucasian populations. CF arises from mutations of the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. The CFTR ion channel orchestrates gating of chloride and bicarbonate ions across epithelial cell membranes in various tissues, including the lung, pancreas, intestine, reproductive tract, and sweat glands. While CF is a systemic disorder, the primary mortality derives from reduced CFTR activity in the airways. Subsequent acidification3 and dehydration leads to accumulation of a viscous mucus layer, occluding the airways and trapping bacteria, leading to infections, reduced lung function, and ultimately, respiratory failure. The most common CFTR mutation, F508del (Class II, found in 90% of CF patients), impairs folding of the CFTR protein (a Class II trafficking defect), resulting in a reduced amount of channel present at the plasma membrane. With the G551D mutation (class III), theamount of protein at the membrane is unaffected, but its open probability (Po) is reduced, also resulting in a reduced channel gating. Thus, to address the underlying causes of CF, two distinct CFTR modulators are required: correctors to increase CFTR levels at the plasma membrane and potentiators to enable effective opening of the channel
Chronic obstructive pulmonary disease (COPD) is anticipated to shortly become the third leading cause of death globally. COPD is characterized by persistent airflow obstruction with cigarette smoke exposure recognized as the primary risk factor. Airflow limitation is associated with all COPD patients; however, the disease is heterogeneous, with variable phenotypes ranging from chronic bronchitis (CB) to emphysema. Small airway disease exhibits increased numbers of goblet cells and mucus plugging with associated smooth muscle hyperplasia, airway fibrosis, and increased inflammation. Excess mucus secretion is believed to play an important role in COPD pathogenesis and is associated with progression of the disease.
Cystic fibrosis (CF) is a fatal genetic disease caused by mutations in the gene encoding the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), a protein kinase A activated epithelial anion channel involved in salt and fluid transport in multiple organs, including the lung. Most CF mutations either reduce the number of CFTR channels at the cell surface (e.g. synthesis or processing mutations) or impair channel function (e.g. gating or conductance mutations) or both.
PCT publication No. WO 2011/113894 describes compounds which restore or enhance the function of mutant and/or wild type CFTR for the treatment of cystic fibrosis, primary ciliary dyskinesia, chronic bronchitis, chronic obstructive pulmonary disease, asthma and other CFTR related diseases. The compounds described therein include (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (Example 5 of WO 2011/113894).
The synthesis described in WO 2011/113894 to make (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide is long, uses expensive starting materials and toxic reagents. Schemes 1 and 2 outline a synthesis from WO 2011/113894 used to make(S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide.
In Scheme 1, the intermediate ethyl 3-amino-5-(trifluoromethyl)picolinate (B4) is made via a Buchwald-Hartwig coupling reaction which requires the use of an expensive starting material (B1) and an expensive palladium catalyst which has to be controlled in the final product. Also, the conversion of B4 to B5 requires the use of NBS, a mutagenic reagent which has to be controlled in the API.
Moreover, the conversion of B5 to B8 is accomplished through the addition of 2,5-hexanedione, a well-known neurotoxin, as shown in Scheme 2. Transformation of the pyrrole in B8 to the amine B9 uses hydroxylamine which is a mutagenic and thermally unstable compound that is dangerous to use in large quantities. The overall process described in WO 2011/113894 requires many protecting group manipulations that lead to a low atom economy and afford a lot of waste. Thus there is a need for an improved synthetic process for making (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide.
PATENT
WO 2018116139,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018116139&_cid=P21-L7D5PQ-39961-1
xample 1: 3-Bromo-6-methoxy-5-(trifluoromethyl)picolinic acid
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (1.4 kg, 5.47 mol), tetramethyl ethylene diamine (TMEDA) (1.75 kg, 15 mol) and tetrahydrofuran (THF) (10kg) were charged to a dry and inert reactor. At -25°C a solution of 2,2,6,6-tetramethyl-piperidinylmagnesium chloride lithiumchloride complex, 1 M in THF/toluene (TMPMgCl.LiCl)(14.5 kg, 15 mol) was slowly added. After stirring the reaction mixture for 30 min., CO2 gas was carefully bubbled into the reactor so that the temperature of the exothermic reaction did not exceed -20°C. The reaction mixture was then quenched onto a mixture of t-butyl methyl ether (TBME) and 5% aq. H2SO4 (50 kg). The biphasic mixture was separated and the organic phase was extracted with 2M NaOH solution. The aqueous phase was acidified to pH 1-2 with 5% aq. H2SO4 and extracted with TBME. After a distillative solvent change to cyclohexane the product was crystallized from cyclohexane to yield 1.1 kg 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid (65% yield).
1H NMR (400 MHz, CDCl3): δ ppm 8.24 (d,J = 0.7Hz, 1 H), 4.12 (s, 3H)
13C NMR (101 MHz, DMSO-d6): δ ppm 54.84, 106.37, 114 (m), 117.6/120.3/123.0/125.7 (m), 141.74, 152.43, 158.63, 165.63
HRMS: [M-H]- expected C8H4BrF3NO3, 297.9405; found C8H4BrF3NO3, 297.9337
Example 2: Methyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (5.0 g, 19.53 mmol) was added to a 100 ml reactor followed by toluene (20 ml) and dimethylcarbonate (17.59 g, 195.30 mmol). To the stirred solution at 20 °C was slowly added 2,2,6, 6-tetramethyl-piperidinylmagnesium chloride lithium chloride complex as a 1 M solution in THF/toluene (27.34 ml, 27.34 mmol) within 45 minutes. A sample was taken and diluted in acetic acid for HPLC analysis in order to confirm full conversion of II to the methylester. Within the same vessel 5% aq. H2SO4 (36 ml) was slowly added to the reaction mixture until a pH below 2 was obtained (caution, exothermic). The biphasic mixture was separated and the lower aqueous phase back-extracted with toluene (10 ml).
In order to isolate the methylester the organic phases were combined and concentrated by rotary evaporation to yield a residue which was chromatographed on reverse-phase silica to yield the final product: methyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate as a yellow solid, 5.3 g, 86 % yield. The solid was optionally recrystallized from methanol and water to further increase purity.
1H NMR (400MHz, CDCl3): δ ppm 8.08 (br s, 1 H), 4.07 (s, 3H), 4.02 (s, 3H)
13C NMR (CDCl3): δ ppm 164.76, 159.22, 149.90, 141.49, 122.83, 120.12, 116.12, 108.05, 54.93, 53.09
HRMS: MH+ expected C9H8BrF3NO3, 313.9561 ; found C9H8BrF3NO3, 313.9634
HPLC Conditions:
HPLC: Column : Agilent Zorbax SB-C18 (150 mm x 3.0 mm, particle size 3.5 urn)
Eluent A : Water / TFA = 1000/1 (v/v)
Eluent B: Acetonitrile / TFA = 1000/1 (v/v)
Wavelength : 230 nm
Flow-rate : 0.8 ml/min
Gradient: eluent B: 45% to 90% over 9 mins
Retention time 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate: 5.80 min
Alternative synthesis for 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid:
Isolation of Example 1
In order to proceed to Example 1 without the isolation of VII, the work-up continues from the combined toluene phases post-H2SO4 quench as follows:
To the combined organic phases was slowly added 50% aq. sodium hydroxide (30 ml) until a pH of above 10 was obtained. The reaction mixture was heated to 35 °C and after 15 mins addition of water (30 ml) followed by 30 mins further stirring preceded sample-taking to ensure full hydrolysis of the methylester to Example 1 by HPLC. Water was added (130 ml), followed by TBME (60 ml) and the phases separated. To the aqueous phase was cautiously added concentrated H2SO4 (30 g) until a pH of below 2.5 was obtained (caution, exothermic and release of CO2 causes foaming). TBME (100 ml) was added and the phases separated. The organic phase contained the C2, and could be evaporated to dryness by rotary evaporation to confirm the yield, 5.4 g C2, 92 % yield.
1H NMR (400 MHz,CDCl3): δ ppm 8.24 (d,J=0.7Hz, 1 H), 4.12 (s, 3H)
13C NMR (101 MHz,DMSO-d6): δ ppm 54.84, 106.37, 114 (m), 117.6/120.3/123.0/125.7 (m), 141.74, 152.43, 158.63, 165.63
HRMS: M-H- expected C8H4BrF3NO3, 297.9405; found C8H4BrF3NO3, 297.9333
For HPLC method details see above. Retention time C2: 2.94 min
Alternative synthesis for ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate:
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (0.5 g, 1.95 mmol) was added to a reactor followed by THF (2 ml) and the solution cooled to 0 °C. To the mixture was added 2,2,6,6-tetramethyl-piperidinylmagnesium chloride lithium chloride complex as a 1 M solution in THF/toluene (4.88 ml, 3.91 mmol), and the mixture was left to stir for 15 minutes at 0 °C. An aliquot of the solution (50 ul) was then added to a reactor containing diethylcarbonate (20 ul, 19.5 mmol). A second aliquot (50 ul) was taken of the metallated II and added to a reactor containing ethyl chloroformate (14 ul, 19.5 mmol). After 2 minutes both reactors were quenched with a 1 :1 mixture of acetonitrile/HCl (1 M). The reaction with diethylcarbonate gave 56 A% of ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate and the reaction with ethyl chloroformate gave 68 A% of ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate product according to the HPLC method described above.
Example 3: Synthesis of (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide
Step 1: 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid (1.3 kg, 4.33 mol) and
copper(II)sulfate pentahydrate (0.108 kg, 0.433 mol) were charged into an inert autoclave
followed by aqueous ammonia 25% (12 kg). The mixture was stirred and heated up to 100 °C, whereby a pressure of 7 bar resulted. The solution was stirred for 2 hr and then cooled down to
5 °C. Sulfuric acid (8 M) was dosed upon cooling, so that a temperature range of 5 °C to 30 °C was held until a pH of about 5 was reached. Isopropylacetate was added and the pH was
further adjusted to 1-2. The phases were separated and the organic phase was azeotropically dried by partial distillation. n-Heptane was added and the mixture stirred for 15 hr at 20 °C
during which the product crystallized out. After filtration and drying 3-amino-6-methoxy-5-(trifluoromethyl)picolinic acid was obtained as a yellow solid (0.92 kg, 90%).
1H NMR (400 MHz, DMSO-d6): δ ppm 7.70 (s, 1 H), 3.89 (s, 3H)
13C NMR (101 MHz, DMSO-d6): δ ppm 53.59, 116.76 m, 123.27, 126.36-117.40 m, 128.04, 142.56, 148.65, 167.62
Step 2: 3-amino-6-methoxy-5-(trifluoromethyl) picolinic acid (20 g, 84.7 mmol) and HATU (38.6 g, 101.6 mmol) were charged to a reactor followed by a solution of (S)-3-amino-1 ,1 ,1-trifluoro-2- methylpropan-2-ol in isopropylacetate (7 %, 188 g, 93 mmol). The solution was stirred at room temperature, diisopropyl ethyl amine (21.9 g, 169 mmol) was added and stirring was continued for at least 16h at 25 °C. Water (250 ml) was then added dropwise within 15 min. keeping the temperature below 25 °C. The water phase was separated and the organic phase was extracted with 5% aqueous HCl , 5% potassium carbonate solution, and water. The organic layer was concentrated to about 60% solution. At 50 °C n-heptane (41 g) was added and the solution was cooled by a linear ramp to 5 °C while adding more n-heptane (131 g). The precipitate was filtered off and dried at 50 °C resulting in a yellow to beige product (S)-3-amino-6-methoxy-N- (3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (21.1 g, 69 % yield).
1H NMR (400 MHz, DMSO-d6): δ ppm 8.30 (m,1 H), 7.68 (s,1 H), 6.69 (s,2H), 6.29(s,1 H), 3.93(s,3H), 3.7-3.4(m,2H), 1.26(s,3H)
13C NMR (101 MHz, DMSO-d6): δ ppm 18.92, 42.15, 53.52, 72.40, 115.5-116.5 m, 118-126 m, 122-130.7 m, 124.82, 128.3 m, 140.95, 148.49, 166.27
Example 4: Telescoped process for the synthesis of the HCl salt of 3-amino-6-methoxy- 5-(trifluoromethyl)picolinic acid (V)
1 Equivalent* of (III) and 6 equivalents of dimethyl carbonate (DMC) were dissolved in 3.5 parts** of toluene at room temperature. To this solution 1.5 equivalent of TMPMgCl.LiCl solution in THF was added at 15-25°C within ca. 1 h. Tert butyl methyl ether (MTBE, 5.9 parts) was added and the mixture was quenched in 7.3 parts of 10% sulfuric acid at 25-40°C. The water phase was discarded and to the organic phase 6.2 parts of 30% sodium hydroxide solution were added. The mixture was stirred well at 40°C for 1-2h. After the successful conversion of (VIII) to (IV), 2.5 parts of water were added to dissolve the partially precipitated sodium carbonate. The water phase was discarded and the organic phase was cooled to 20°C and extracted with 4.8 parts of 25% aqueous ammonia. The aqueous phase was transferred in an autoclave and 0.0979 parts (10mol%) of copper sulfate pentahydrate were added. The autoclave was well inertized by a pressure method and heated up to 100°C, while the pressure raises up to ca. 8 bar absolute pressure. After the successful conversion of (IV) to (V), the green solution was added to a mixture of 3.7 parts of MTBE and 6.8 parts of 50% sulfuric acid resulting in a biphasic solution of pH 1-2. The water phase was separated and the organic phase washed two times with 2.5 parts of water each. The organic phase was dried by distillation at JT 50°C/400mbar while 3.7 parts of MTBE were added/replaced. To the dried organic solution 0.41 parts of HCl gas was dosed at 0-5°C under or over solvent level. The suspension was stirred for ca.1 h, then filtered off and washed with 48 parts of TBME. The product was dried at 40°C/20 mbar for ca. 12h. (yield from (III): 72%, slightly beige solid).
*equivalents are based on the molar amount of the starting material (III) = 1 equivalent
**parts = weight/weight (III)
1H NMR (400 MHz, DMSO-d6): δ ppm 7.70 (s, 1 H), 3.89 (s, 3H)
13C NMR (101 MHz, DMSO-d6): δ ppm 53.59, 116.76 m, 123.27, 126.36-117.40 m, 128.04, 142.56, 148.65, 167.62
Example 5: Alternative synthesis of (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide
Step 1 : (VIII) (1.0 g), (S)-3-amino-1 ,1 ,1-trifluoro-2-methylpropan-2-ol as mandellic acid salt (1.128 g, 1.2 eq.) and 2,3,4,6, 7, 8-hexahydro-1H-pyrimido[1,2-a]pyrimidine (TBD, 0.588 g, 1.3 eq.) were added to a pre-dried flask as solids. To this was added the anhydrous THF (10 ml) and the cloudy solution heated to 55 °C. Sampling and analytical determination of purity at 2.5 hrs confirmed 88 A% product upon which water (10 ml) was added and the phases separated. The organic phase was distilled to a concentrated mixture upon which toluene (20 ml) was added. The organic layer was extracted with 10% aq. citric acid (10 ml) followed by three consecutive extractions with 1 M aq. NaOH. The organic phase was then dried with magnesium sulfate and evaporated to dryness to give 1.196 g of (S)-3-bromo-6-methoxy-N-(3,3,3-trifluoro- 2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (IX) as a white solid (95 A%, 88% yield).
1H NMR, CDCl3: δ ppm 8.08 (s, 1 H), 7.83 (br s, 1 H), 3.99 (s, 3H), 3.78-3.60 (m, 2H), 3.51 (br s, 1 H), 1.36 (s, 3H)
19F NMR, CDCl3: δ ppm -64.28, -81.44
13C DEPT135, CDCl3: δ ppm 144.20 (CH), 54.70 (CH3), 44.26 (CH2), 19.71 (CH3)
HRMS: MH+ expected C12H12BrF6N2O3, 424.9857; found C12H12BrF6N2O3, 424.9931
HPLC (method described above): retention time = 4.94 min
Step 2: IX (79 mg, 0.186 mmol) was combined with copper(II)sulfate pentahydrate (4.6 mg, 0.019 mmol), methanol (0.6 ml) and 23% aqueous ammonium hydroxide solution (559 ul) within a glass microwave vial. The headspace was inertized with nitrogen, then the vial sealed and placed in the microwave unit for heating to 105 °C for 7.5 hrs. Isopropylacetate (5 ml) was added to the deep green reaction mixture and a solvent-switch brought about by rotary evaporation. To the mixture now in water and isopropyl acetate was added 8M H2SO4 (5 ml), the phases mixed and then left to separate. The aqueous phase was further extracted with isopropylacetate and the combined organic phases washed with aq. NaCl (5 ml). The organic phase was dried over MgSO4 and evaporated to yield of a yellow residue, 66 mg.
A portion of the residue (16 mg) was re-dissolved in heptane / ethyl acetate and submitted for combiflash purification (n-heptane / ethyl acetate gradient, elution at 20% ethyl acetate) providing (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (VII) as a residue on evaporation in 91 A% purity containing trace residual solvents (17 mg, corrected to 13 mg by 1H NMR, 80 % yield back-calculated).
1H NMR, CDCl3: δ ppm 8.11 (br s, 1 H), 7.37 (s, 1 H), 3.97 (s, 3H), 3.76-3.72 (d, 2H, J=6.3Hz), 1.42 (s, 3H)
13C NMR, CDCl3: δ ppm 168.86, 150.55, 140.21 , 128.63, 127.26, 125.35, 124.42, 123.39, 120.68, 118.60, 74.16, 53.73, 44.39, 19.55
ESI-MS: expected mass 361.2. ELS detector, 100 A%, MH+ 362.1 , M- 360.1
HPLC (method described above): retention time = 4.39 min
PATENT
US20200383960
https://patentscope.wipo.int/search/en/detail.jsf?docId=US312969607&_cid=P21-L7D5H8-38258-1
Examples 4, 5 and 6: 3-Amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid (3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide and its enantiomers
was prepared according to the following procedure:
Examples 5 and 6 are Entantiomers
SYN
J. Med. Chem. 2021, 64, 11, 7241–7260
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) ion channel are established as the primary causative factor in the devastating lung disease cystic fibrosis (CF). More recently, cigarette smoke exposure has been shown to be associated with dysfunctional airway epithelial ion transport, suggesting a role for CFTR in the pathogenesis of chronic obstructive pulmonary disease (COPD). Here, the identification and characterization of a high throughput screening hit 6 as a potentiator of mutant human F508del and wild-type CFTR channels is reported. The design, synthesis, and biological evaluation of compounds 7–33 to establish structure–activity relationships of the scaffold are described, leading to the identification of clinical development compound icenticaftor (QBW251) 33, which has subsequently progressed to deliver two positive clinical proofs of concept in patients with CF and COPD and is now being further developed as a novel therapeutic approach for COPD patients.

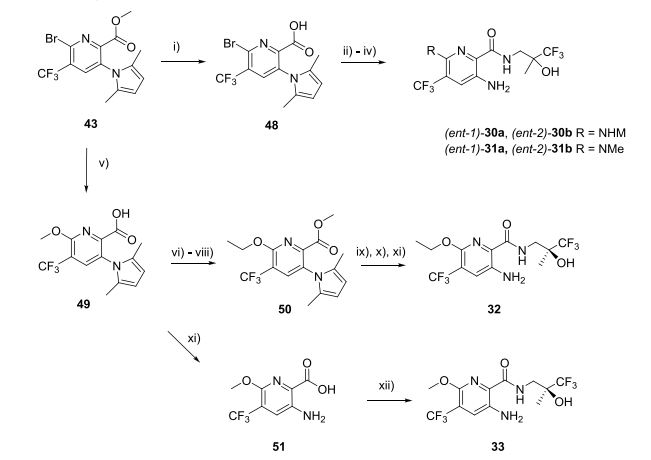
a Reagents and conditions: (i) aq NaOH, THF, RT, 97%; (ii) aq Me2NH or MeNH2, THF, RT, 56−92%; (iii) 41, HATU, Et3N, NMP, RT, 52− 78%; (iv) NH2OH·HCl, Et3N, EtOH−water, reflux, then chiral HPLC, 34−36%; (v) aq NaOH, MeOH, 60°C, 97%; (vi) cat H2SO4, MeOH, reflux, 75%; (vii) TMSCl, KI, MeCN, reflux, 54%; (viii) EtOH, DEAD, Ph3P, dioxane, RT, 61%; (ix) aq NaOH, THF, reflux, 26%; (x) (S)-41, HATU, DIPEA, DMF, RT, 89%; (xi) NH2OH·HCl, Et3N, EtOH−water, reflux, 37−53%; (xii) (S)-41, HATU, DIPEA, NMP, RT, 59%.
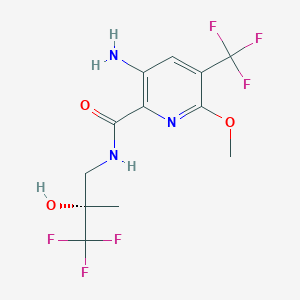
(S)-3-Amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2- methylpropyl)-5-(trifluoromethyl)picolinamide
(S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2- methylpropyl)-5-(trifluoromethyl)picolinamide 33 as a white solid (33.6 g, 59%). LRMS C12H13F6N3O3 requires M+ 361.08, found [MH]+ 362.2. Elemental analysis requires C, 39.90%; H, 3.63%; N, 11.63% found C, 40.22 ± 0.06%; H, 3.68 ± 0.11%; N, 11.76 ± 0.04%. 1 H NMR (DMSO-d6) 1.26 (3H s), 3.46 (1H dd J = 13.3, 5.6), 3.66 (1H dd J = 13.7, 7.3), 3.92 (3H s), 6.29 (1H s), 6.69 (2H br s), 7.68 (1H s), 8.30 (1H t J = 6.4). 13C NMR (DMSO-d6) 18.95 (q), 42.19 (t), 53.56 (q), 72.27 (s JF = 26.8), 116.07 (s JF = 32.3), 122.40 (s JF = 272.1), 124.85 (s), 126.43 (s JF = 287.1), 128.29 (d JF = 5.2), 141.0 (s), 148.51 (s), 166.3 (s). 19F NMR (DMSO-d6) −62.71 (s), −80.46 (s).
/////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Other names | QBW251 |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C12H13F6N3O3 |
| Molar mass | 361.244 g·mol−1 |
| 3D model (JSmol) | |
References
- ^ Rowe SM, Jones I, Dransfield MT, Haque N, Gleason S, Hayes KA, et al. (2020). “Efficacy and Safety of the CFTR Potentiator Icenticaftor (QBW251) in COPD: Results from a Phase 2 Randomized Trial”. International Journal of Chronic Obstructive Pulmonary Disease. 15: 2399–2409. doi:10.2147/COPD.S257474. PMC 7547289. PMID 33116455.
- ^ Grand DL, Gosling M, Baettig U, Bahra P, Bala K, Brocklehurst C, et al. (June 2021). “Discovery of Icenticaftor (QBW251), a Cystic Fibrosis Transmembrane Conductance Regulator Potentiator with Clinical Efficacy in Cystic Fibrosis and Chronic Obstructive Pulmonary Disease”. Journal of Medicinal Chemistry. 64 (11): 7241–7260. doi:10.1021/acs.jmedchem.1c00343. ISSN 0022-2623. PMID 34028270.
- ^ Kazani S, Rowlands DJ, Bottoli I, Milojevic J, Alcantara J, Jones I, et al. (March 2021). “Safety and efficacy of the cystic fibrosis transmembrane conductance regulator potentiator icenticaftor (QBW251)”. Journal of Cystic Fibrosis. 20 (2): 250–256. doi:10.1016/j.jcf.2020.11.002. PMID 33293212.
- ^ Ray F (December 9, 2020). “Icenticaftor Effective in CF Patients With Certain Mutations, Phase 1/2 Trial Shows”. cysticfibrosisnewstoday.com. BioNews Services.
- ^ Jump up to:a b “Icenticaftor – Novartis”. Adis Insight. Springer Nature Switzerland AG.
////////////QBW 251, ICENTICAFTOR, NOVARTIS, chronic obstructive pulmonary disease, COPD, cystic fibrosis,
C[C@](CNC(=O)C1=C(C=C(C(=N1)OC)C(F)(F)F)N)(C(F)(F)F)O