















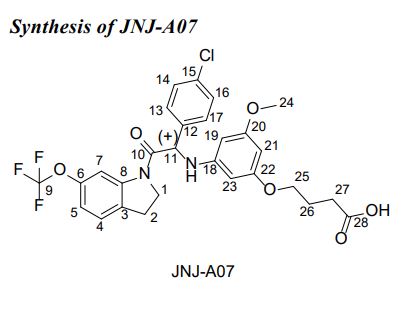
JNJ-A07
S + FORM
CAS 2135640-93-4 ROT (+)S
Butanoic acid, 4-[3-[[1-(4-chlorophenyl)-2-[2,3-dihydro-6-(trifluoromethoxy)-1H-indol-1-yl]-2-oxoethyl]amino]-5-methoxyphenoxy]-, (+)-
(+)-4-[3-[[1-(4-Chlorophenyl)-2-[2,3-dihydro-6-(trifluoromethoxy)-1H-indol-1-yl]-2-oxoethyl]amino]-5-methoxyphenoxy]butanoic acid
(+)-4-[3-([(1S)-1-(4-Chlorophenyl)-2-oxo-2-[6-(trifluoromethoxy)-2,3-dihydro-1H-indol-1-yl]ethyl]amino)-5-methoxyphenoxy]butanoic acid
REF
Kaptein, S.J.F., Goethals, O., Kiemel, D. et al. A pan-serotype dengue virus inhibitor targeting the NS3–NS4B interaction. Nature (2021). https://doi.org/10.1038/s41586-021-03990-6

JNJ-018
CAS 2135640-91-2 +/-, R,S
CAS 2135640-92-3 ROT (-)R
Butanoic acid, 4-[3-[[1-(4-chlorophenyl)-2-[2,3-dihydro-6-(trifluoromethoxy)-1H-indol-1-yl]-2-oxoethyl]amino]-5-methoxyphenoxy]-, (-)-
(-)-4-[3-[[1-(4-Chlorophenyl)-2-[2,3-dihydro-6-(trifluoromethoxy)-1H-indol-1-yl]-2-oxoethyl]amino]-5-methoxyphenoxy]butanoic acid
- Janssen (Originator)
- Katholieke Universiteit Leuven (Originator)
- NS4B Protease (Dengue Virus) Inhibitors
- Serine Protease NS3/Non-Structural Protein NS4B Protease (Dengue Virus) Interaction Inhibitors
A pan-serotype dengue virus inhibitor targeting the NS3–NS4B interaction
https://www.nature.com/articles/s41586-021-03990-6
https://www.nature.com/articles/s41586-021-03990-6#citeas
Abstract
Dengue virus causes approximately 96 million symptomatic infections annually, manifesting as dengue fever or occasionally as severe dengue1,2. There are no antiviral agents available to prevent or treat dengue. Here, we describe a highly potent dengue virus inhibitor (JNJ-A07) that exerts nanomolar to picomolar activity against a panel of 21 clinical isolates that represent the natural genetic diversity of known genotypes and serotypes. The molecule has a high barrier to resistance and prevents the formation of the viral replication complex by blocking the interaction between two viral proteins (NS3 and NS4B), thus revealing a previously undescribed mechanism of antiviral action. JNJ-A07 has a favourable pharmacokinetic profile that results in outstanding efficacy against dengue virus infection in mouse infection models. Delaying start of treatment until peak viraemia results in a rapid and significant reduction in viral load. An analogue is currently in further development.
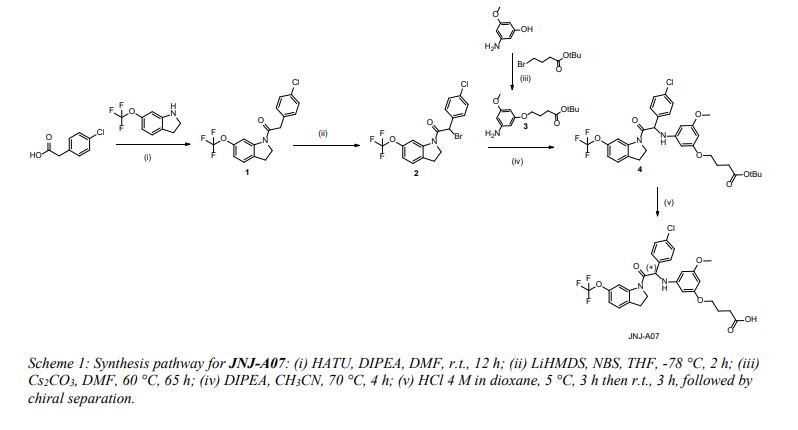
2-(4-Chlorophenyl)-1-(6-(trifluoromethoxy)indolin-1-yl)-ethanone (1)
127 A mixture of 6-(trifluoromethoxy)indoline ([CAS 959235-95-1], 2 g, 9.84 mmol), 2-(4-chlorophenyl)acetic acid 128 ([CAS 1878-66-6], 1.85 g, 10.8 mmol), HATU (5.6 g, 14.8 mmol) and diisopropylethylamine (4.9 mL, 29.5
129 mmol) in DMF (40 mL) was stirred at room temperature for 12 h. Water was added and the precipitate was
130 filtered off. The residue was taken up with EtOAc. The organic solution was washed with a 10 % aqueous
131 solution of K2CO3, brine, dried over MgSO4, filtered, and the solvent was evaporated under reduced pressure. 132 The residue was purified by chromatography on silica gel (15-40 pm, 80 g, heptane/EtOAc gradient 90/10 to 133 60/40). The pure fractions were combined and the solvent was concentrated under reduced pressure to give 2-(4-
134 chlorophenyl)-1-(6-(trifluoromethoxy)indolin-1-yl)-ethanone 1 (3 g, yield: 86 %).
135 1 H NMR (400 MHz, DMSO-d6) d ppm 7.99 (s, 1 H), 7.37 – 7.41 (m, 2 H), 7.29 – 7.34 (m, 3 H), 6.97 (dd, J = 8.1, 1.3 Hz, 1 H), 4.25 (t, J = 8.6 Hz, 2 H), 3.88 (s, 2 H), 3.18 (t, J = 8.5 Hz, 2 H); 13
136 C NMR (101 MHz, 137 CHLOROFORM-d) δ ppm 168.91, 148.65, 148.63, 144.05, 133.16, 132.26, 130.63, 129.54, 128.93, 124.87, 120.50 (q, J=257.2 Hz), 116.38, 110.83, 77.26, 48.86, 42.52, 27.59; LC-MS: [M+H]+
138 728; purity 99 % (method LCMS2); Melting Point: 116-131 °C (DSC peak: 120.2 °C); HRMS (ESI+) m/z: [M]+ 139 calcd for C17H13ClF3NO2,
140 356.0660; found, 356.0657
141 2-Bromo-2-(4-chlorophenyl)-1- (6-(trifluoromethoxy)indolin-1-yl)ethanone (2)
142 At -78 °C, under nitrogen flow, LiHMDS (1.5 M in THF, 11.2 mL, 16.9 mmol) was added dropwise to a mixture 143 of 1 (3 g, 8.43 mmol) in THF (50 mL). The mixture was stirred for 15 min at -78 °C and a solution of N
144 bromosuccinimide (1.65 g, 9.3 mmol) in THF (30 mL) was added dropwise. After stirring for 2 h at -78 °C, the 145 reaction was quenched with a saturated aqueous solution of NH4Cl. The mixture was extracted with EtOAc. The 146 organic layer was separated, dried over MgSO4, filtered, and the solvent was evaporated under reduced pressure
147 to give 2-bromo-2-(4-chlorophenyl)-1- (6-(trifluoromethoxy)indolin-1-yl)ethanone 2 (3.6 g, yield: 98 %) as an 148 oil. The compound was used without further purification in the next step.
149 1 H NMR (400 MHz, CHLOROFORM-d) δ ppm 8.19 (s), 7.52 – 7.57 (m), 7.34 – 7.39 (m), 7.17 (d, J=8.2 Hz), 6.92 (dd, J=8.2, 1.1 Hz), 5.56 (s), 4.37 (td, J=10.1, 6.5 Hz), 4.09 (td, J=10.1, 6.7 Hz), 3.12 – 3.31 (m); 13
150 C NMR
151 (101 MHz, CHLOROFORM-d) δ ppm 164.90 (s), 148.68 (d, J=2.2 Hz), 143.75 (s), 135.46 (s), 133.99 (s), 152 130.52 (s), 129.79 (s), 129.10 (s), 125.01 (s), 117.20 (s), 120.47 (q, J=257.2 Hz), 111.36 (s), 48.88 (s), 46.61 (s), 27.65 (s); LC-MS: [M+H]+ 436; purity 100 % (method LCMS2); HRMS (ESI+) m/z: [M]+ 153 calcd for
154 C17H13O2NBrClF3, 433.9765; found, 433.9764
155 tert-Butyl 4-(3-amino-5-methoxyphenoxy)butanoate (3) 156 To a mechanically stirred solution of tert-butyl 4-bromobutanoate ([CAS 110661- 5 91-1], 42.3 g, 0.19 mol) in
157 DMF (600 mL) was added in portions a solid mixture of 3-amino-5-methoxyphenol ([CAS 162155-27-3], 26.4 158 g, 0.19 mol) and Cs2CO3 (123.6 g, 0.379 mol). The reaction mixture was stirred at 60 °C for 65 h, and allowed to
159 reach room temperature. The mixture was poured out into water (2.5 L). The product was extracted with Et2O (2 160 x). The combined organic layers were washed with brine, dried over MgSO4, and filtered. The solvent was
161 evaporated under reduced pressure, and then co-evaporated with toluene. The residue was purified by normal 162 phase HPLC (Stationary phase: silica gel 60A 25-40 pm (Merck), Mobile phase: gradient EtOAc/heptane 20/80 163 to 60/40), yielding tert-butyl 4-(3-amino-5-methoxyphenoxy)butanoate 3 as an oil (27 g, yield: 50 %).
164 1 H NMR (400 MHz, CHLOROFORM-d) δ ppm 5.89 – 5.92 (m), 5.86 (d, J=2.2 Hz), 3.92 (t, J=6.2 Hz), 3.73 (s), 3.66 (br s), 2.40 (t, J=7.4 Hz), 1.98 – 2.08 (m), 1.45 (s); 13
165 C NMR (101 MHz, CHLOROFORM-d) δ ppm 172.61 166 (s), 161.69 (s), 161.02 (s), 148.35 (s), 94.33 (s), 93.89 (s), 91.52 (s), 80.35 (s), 66.74 (s), 55.17 (s), 32.07 (s), 28.13 (s), 24.78 (s); LC-MS: [M+H]+ 282; purity 94 % (method LCMS2); HRMS (ESI+) m/z: [M]+
167 calcd for 168 C15H24O4N, 282.1700; found, 282.1695
169 tert-Butyl 4-(3-((1-(4-chlorophenyl)-2-oxo-2-(6-(trifluoromethoxy)indolin-1-yl)ethyl)amino)-5-
170 methoxyphenoxy)butanoate (4)
171 A mixture of 2 (3.6 g, 8.3 mmol), 3 (2.3 g, 8.3 mmol) and diisopropylethylamine (1.7 ml, 9.94 mmol) in CH3CN 172 (80 mL) was stirred at 70 °C for 4 h. The mixture was concentrated under reduced pressure, diluted with EtOAc,
173 and washed with 1 N aqueous HCl and water. The organic phase was separated, dried over MgSO4, filtered, and 174 the solvent was evaporated under reduced pressure. The compound was purified by flash chromatography on 175 silica gel (15-40 pm, 120 g, heptane/EtOAc 80/20). The pure fractions were combined and evaporated to dryness
176 to give, after crystallization from diisopropyl ether, tert-butyl 4-(3-((1-(4-chlorophenyl)-2-oxo-2-(6-
177 (trifluoromethoxy)indolin-1-yl)ethyl)amino)-5-methoxyphenoxy)butanoate 4 (2.6 g, yield: 49 %).
178 1 H NMR (400 MHz, DMSO-d6) d ppm 8.03 (s, 1 H), 7.55 (d, J = 8.6 Hz, 2 H), 7.43 (d, J = 8.6 Hz, 2 H), 7.33 (d, 179 J = 8.1 Hz, 1 H), 7.01 (dd, J = 8.1, 1.5 Hz, 1 H), 6.44 (d, J = 8.8 Hz, 1 H), 5.94 (d, J = 2.0 Hz, 2 H), 5.75 (t, J = 180 2.0 Hz, 1 H), 5.55 (d, J = 8.8 Hz, 1 H), 4.51 (td, J = 10.3, 6.5 Hz, 1 H), 4.04 (td, J = 10.3, 7.3 Hz, 1 H), 3.84 (t, J 6 181 = 6.3 Hz, 2 H), 3.62 (s, 3 H), 3.09 – 3.23 (m, 2 H), 2.31 (t, J = 7.3 Hz, 2 H), 1.86 (quin, J = 6.8 Hz, 2 H), 1.39 (s, 9 H); 13
182 C NMR (101 MHz, CHLOROFORM-d) δ ppm 172.57, 168.84, 161.66, 161.02, 148.65, 148.63, 147.68,
183 143.79, 135.66, 134.48, 129.58, 129.42, 129.38, 124.99, 116.92, 120.50 (q, J=257.2 Hz), 111.13, 93.02, 92.72, 91.06, 80.38, 77.25, 66.79, 59.74, 55.17, 48.31, 32.09, 28.15, 27.64, 24.77; LC-MS: [M+H]+
184 635; purity: 98 % (method LCMS3); Melting Point: 109-125 °C (DSC peak: 116.1 °C); HRMS (ESI+) m/z: [M]+ 185 calcd for 186 C32H34ClF3N2O6, 635.2130; found, 635.2127 187 (+)-4-(3-((1-(4-Chlorophenyl)-2-oxo2-(6-(trifluoromethoxy)indolin-1-yl)ethyl)amino)-5-
188 methoxyphenoxy)butanoic acid (JNJ-A07) 189 A solution of 4 (2.4 g, 3.8 mmol) in 4 M HCl in dioxane (24 mL) was stirred at 5 °C for 3 h and at room 190 temperature for 3 h. The precipitate was filtered off and dried to afford 4-(3-((1-(4-chlorophenyl)-2-oxo2-(6- 191 (trifluoromethoxy)indolin-1-yl)ethyl)amino)-5-methoxyphenoxy)butanoic acid as an HCl salt (racemic JNJ192 A07, 2 g, 0.8 eq. HCl, 0.07 eq. H2O). This salt was neutralized prior to chiral separation by dissolving it in 193 EtOAc and treating this solution with 1 N aqueous NaOH and evaporation of the organic layer under reduced 194 pressure.
The enantiomers were separated via preparative chiral SFC (Stationary phase: Chiralcel® OD-H 5 pm 195 250 x 30 mm, Mobile phase: 50 % CO2, 50 % iPrOH (+ 0.3 % iPrNH2)) and further purified via preparative 196 achiral SFC (Stationary phase: Cyano® 6 pm 150 x 21.2 mm, Mobile phase: 80 % CO2, 20 % MeOH (+ 0.3 % 197 iPrNH2)). The product fractions were combined and evaporated under reduced pressure. Each enantiomer was 198 taken up with EtOAc and washed with 1 N aqueous HCl. The organic layers were separated, dried over MgSO4, 199 filtered, and the solvent was evaporated under reduced pressure. The first eluted enantiomer was solidified from 200 diethyl ether/diisopropyl ether to give the epimer of JNJ-A07 (616 mg, yield: 28 %).
The second eluted
201 enantiomer was solidified from diethyl ether/diisopropyl ether to give JNJ-A07 (715 mg, yield: 32 %).
202 203 Epimer of JNJ-A07:
204 1 H NMR (500 MHz, DMSO-d6) δ ppm 12.12 (br s, 1 H), 8.04 (br s, 1 H), 7.55 (br d, J = 8.2 Hz, 2 H), 7.44 (br d, 205 J = 8.5 Hz, 2 H), 7.34 (br d, J = 7.9 Hz, 1 H), 7.01 (br d, J = 7.6 Hz, 1 H), 6.45 (br s, 1 H), 5.95 (br d, J = 10.1 206 Hz, 2 H), 5.76 (s, 1 H), 5.57 (br s, 1 H), 4.47 – 4.57 (m, 1 H), 3.99 – 4.11 (m, 1 H), 3.85 (br t, J = 6.3 Hz, 2 H), 3.62 (s, 3 H), 3.08 – 3.27 (m, 2 H), 2.34 (br t, J = 7.3 Hz, 2 H), 1.87 (quin, J = 6.7 Hz, 2 H); 13
207 C NMR (101 MHz, 208 DMSO-d6) δ ppm 174.56, 169.79, 161.46, 160.71, 149.08, 147.64, 144.48, 137.28, 132.91, 131.95, 130.56, 209 128.89, 126.34, 120.58 (d, J=256.0 Hz), 116.69, 109.52, 93.08, 92.80, 90.23, 66.65, 58.69, 55.20, 48.65, 30.57, 27.48, 24.72; LC-MS: [M+H]+ 579; purity: 100 % (method LCMS1);
Chiral SFC: [M+H]+ 210 579; chiral purity 100 % (method SFC1); [a]D20 211 : -48.5° (589 nm, c 0.27 w/v %, DMF, 20 °C); Melting Point: 62-80 °C (DSC peak: 70.6 °C); HRMS (ESI+) m/z: [M]+ 212 calcd for C28H27O6N2ClF3, 579.1504; found, 579.1501 213 214
JNJ-A07: 215 1 H NMR (500 MHz, DMSO-d6) δ ppm 12.12 (brs, 1 H), 8.04 (br s, 1 H), 7.55 (br d, J = 8.2 Hz, 2 H), 7.44 (br d, 216 J = 8.2 Hz, 2 H), 7.34 (br d, J = 7.9 Hz, 1 H), 7.01 (br d, J = 7.9 Hz, 1 H), 6.45 (br s, 1 H), 5.95 (br d, J = 10.1 217 Hz, 2 H), 5.76 (br s, 1 H), 5.57 (s, 1 H), 4.46 – 4.59 (m, 1 H), 3.99 – 4.10 (m, 1 H), 3.85 (br t, J = 6.1 Hz, 2 H), 3.62 (s, 3 H), 3.09 – 3.27 (m, 2 H), 2.34 (br t, J = 7.3 Hz, 2 H), 1.87 (br t, J = 6.8 Hz, 2 H); 13
218 C NMR (101 MHz,
219 DMSO-d6) δ ppm 174.53 (C28), 169.79 (C10), 161.47 (C20), 160.72 (C22), 149.08 (C18), 147.65 (C6), 144.48 220 (C8), 137.29 (C12), 132.92 (C15), 131.95 (C3), 130.56 (C13, C17), 128.89 (C14, C16), 126.34 (C4), 120.58 (q, 221 J = 255.1 Hz, C9), 116.67 (C5), 109.51 (C7), 93.11 (C23), 92.81 (C21), 90.26 (C19), 66.66 (C25), 58.70 (C11), 55.21 (C24), 48.67 (C1), 30.57 (C27), 27.49 (C2), 24.72 (C26); LC/MS: [M+H]+
222 579; purity 100 % (method LCMS1); Chiral SFC: [M+H]+ 579; chiral purity 100 % (method SFC1); [a]D20
223 : +42.9° (589 nm, c 0.28 w/v %, 224 DMF, 20 °C); Melting point: 62-78 °C (DSC peak: 71.3 °C) ; HRMS (ESI+) m/z calcd for C28H27O6N2ClF3 [M]+ 225 , 579.1504, found 579.1500; Elemental analysis requires C, 58.09 %; H, 4.53 %; N, 4.84 % found C, 226 58.60 %; H, 4.59 %; N, 4.80 %
CLIP
https://www.bioworld.com/articles/512333-potent-selective-pan-serotype-dengue-inhibitor-developed
Blocking the interaction between two dengue virus (DENV) nonstructural proteins, NS3 and NS4B, with a newly developed small-molecule inhibitor resulted in potent antiviral activity in mouse models, according to an international collaborative study led by scientists at the University of Leuven (KU Leuven), CD3 the Centre for Drug Design and Discovery in Leuven, and Janssen Pharmaceutica in Beerse, Belgium.
This protein interaction represents a promising new target for the development of pan-serotype DENV inhibitors with a high barrier to resistance, with the potency of the inhibition warranting further development of these compounds, the authors reported in the October 6, 2021, edition of Nature.
“This is the first study to show that blocking the NS3/NS4B interaction has potent antiviral activity in mice warranting the further development of such inhibitors,” said study co-leader Johan Neyts, professor of virology at KU Leuven.
Dengue is currently among the leading threats to global public health, with an estimated 96 million individuals developing dengue disease, which is probably an underestimation.
In addition, the incidence of dengue has increased approximately 30-fold over the past 50 years. DENV is now endemic in the subtropical regions of 128 countries, with an estimated 4 billion people at risk of infection, predicted to increase to 6 billion by 2080.
This dengue upsurge is driven by various factors, most notably rapid urbanization and the spread of the Aedes mosquito vectors due to climate change.
The DENV has four serotypes that are further classified into genotypes, which are increasingly co-circulating in endemic regions. Antibodies to infection with one serotype can lead to a more severe second infection with a different serotype increases the risk of potentially life-threatening severe dengue.
The DENV vaccine Dengvaxia (Sanofi-Pasteur), which has been approved in several countries for individuals aged at least 9 years, is only recommended for those with previous DENV exposure.
Moreover, there are currently no available antiviral agents for dengue prevention or treatment, while development of pan-serotype DENV inhibitors has proven challenging.
“The major developmental challenge has been to obtain ultrapotent antivirals that also have equipotent activity against the four DENV serotypes,” Neyts told BioWorld Science.
Such drugs should lower viral loads during an ongoing infection, thereby reducing dengue-associated morbidity and mortality, as well as transmission.
In their new Nature study, researchers co-led by Neyts, Patrick Chaltin, managing director of CD3 the Centre for Drug Design and Discovery, and Marnix Van Loock, R&D Lead Emerging Pathogens, Janssen Global Public Health at Janssen Pharmaceutica, identified potential new DENV inhibitors using large-scale cell-based anti-DENV-2 screening.
“We screened tens of thousands of molecules and interesting hits were further optimized to eventually obtain JNJ-A07 and other ultrapotent and selective analogues, with roughly 2,000 analogues being synthesized and tested,” said Neyts.
Notably, the promising small molecule JNJ-A07 was demonstrated to have nanomolar to picomolar activity against a panel of 21 clinical isolates representing the natural genetic diversity of known DENV genotypes and serotypes.
The molecule was then shown to have a high barrier to resistance “by months of culturing the dengue virus in suboptimal concentrations of the inhibitor,” Neyts said.
JNJ-A07 was then shown to prevent formation of the viral replication complex by blocking the interaction between the nonstructural proteins NS3 and NS4B, thereby revealing a previously undescribed mechanism of antiviral action.
JNJ-A07 was further demonstrated to have a favorable pharmacokinetic (PK) profile resulting in outstanding efficacy against DENV infection in mouse models.
“JNJ-A07’s favorable PK profile resulted from optimization of the ADME [absorption, distribution metabolism and excretion] properties of the analogues within this chemical series,” Janssen’s Van Loock told BioWorld Science.
“This enabled us to administer the compound [twice daily] in mice and assess its efficacy, which resulted in a significantly reduced viral load and protected against mortality in a mouse lethal challenge model.”
However, “additional research will be required in preclinical models, to understand how these findings reflect those in humans, as currently no translational models are available to assess the potential effect in humans,” noted Van Loock.
Delaying treatment commencement until peak viremia had developed was shown to result in a rapid and significant reduction in viral load in the mouse models of infection.
This is an important finding, as “one wants an antiviral effect that is independent of how much [viral] replication is ongoing,” Van Loock said.
“In these mice, the reduction in viral load was also very pronounced if the treatment was initiated on the day of peak viral load, when the effect was quantified 24 hours later.”
On safety, said Neyts, as JNJ-A07 and its analogues “target specific viral proteins that have no homologues in eukaryotic cells, we expect a considerable safety window, with these agents being very well tolerated.” The safety and potency of DENV inhibition established in this study justifies the further development of these novel antivirals, with an analogue being currently in further development.
Further development will include “using our know-how to also develop drugs against the other member of the flavivirus family to which DENV belongs, including Japanese encephalitis, Zika, yellow fever, West Nile virus, et cetera,” said Neyts.
Meanwhile, “Janssen has moved the compound into clinical development and continues to work closely in this regard with teams at KU Leuven and elsewhere,” said Van Loock.
“We will be sharing information about progress of the compound’s clinical development during the American Society of Tropical Medicine and Hygiene meeting this November.”

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
-
IN201827040889
-
US2020299235
-
US2019112266
-
US10689340
Due to the presence of said chiral carbon atom, a “compound of formula (I)” can be the (R)-enantiomer, the (S)-enantiomer, the racemic form, or any possible combination of the two individual enantiomers in any ratio. When the absolute (R)-or (S)-configuration of an enantiomer is not known, this enantiomer can also be identified by indicating whether the enantiomer is dextrorotatory (+)- or levorotatory (-)- after measuring the specific optical rotation of said particular enantiomer.
In an aspect the present invention relates to a first group of compound of formula (I) wherein the compounds of formula (I) have the (+) specific rotation.
In a further aspect the present invention relates to a second ground of compounds of formula (I) wherein the compounds of formula (I) have the (-) specific rotation.
Example 4: synthesis of 4-(3-((1 -(4-chlorophenyl)-2-oxo-2-(6-(trifluoromethoxy)-indolin-1 -yl)ethyl)amino)-5-methoxyphenoxy)butanoic acid (Compound 4) and chiral separation into Enantiomers 4A and 4B.
Synthesis of intermediate 4a:
A mixture of 6-(trifluoromethoxy)indoline [CAS 959235-95-1] (2 g, 9.84 mmol), 2-(4-chlorophenyl)acetic acid [CAS 1878-66-6] (1 .85 g, 10.8 mmol), HATU (5.6 g, 14.8 mmol) and diisopropylethylamine (4.9 ml_, 29.5 mmol) in DMF (40 ml_) was stirred at room temperature for 12 h. Water was added and the precipitate was filtered off. The residue was taken up with EtOAc. The organic solution was washed with a 10% aqueous solution of K2CO3, brine, dried over MgSO4, filtered and the solvent was evaporated under reduced pressure. The residue was purified by chromatography on silica gel (15-40 μιτι, 80 g, heptane/EtOAc gradient 90/10 to 60/40). The pure fractions were combined and the solvent was concentrated under reduced pressure to give 2-(4-chlorophenyl)-1 -(6-(trifluoromethoxy)indolin-1 -yl)-ethanone 4a (3 g).
Synthesis of intermediate 4b:
At -78°C, under N2 flow, LiHMDS 1 .5 M in THF (1 1 .2 ml_, 16.9 mmol) was added dropwise to a mixture of 2-(4-chlorophenyl)-1 -(6-(trifluoromethoxy)indolin-1 -yl)-ethanone 4a (3 g, 8.43 mmol) in THF (50 ml_). The mixture was stirred for 15 min at -78°C and a solution of /V-bromosuccinimide (1 .65 g, 9.3 mmol) in THF (30 ml_) was added dropwise. After stirring for 2 h at -78°C, the reaction was quenched with a saturated solution of NH CI. The mixture was extracted with EtOAc. The organic layer was separated, dried over MgSO4, filtered and the solvent was evaporated under reduced pressure to give 2-bromo-2-(4-chlorophenyl)-1 -(6-(trifluoromethoxy)indolin-1 -yl)ethanone 4b (3.6 g). The compound was used as such in the next step.
Synthesis of intermediate 4c:
A mixture of 2-bromo-2-(4-chlorophenyl)-1 -(6-(trifluoromethoxy)indolin-1 -yl)-ethanone 4b (3.6 g, 8.3 mmol), terf-butyl 4-(3-amino-5-methoxyphenoxy)-butanoate 1a (2.3 g, 8.3 mmol) and diisopropylethylamine (1 .7 mL, 9.94 mmol) in CH3CN (80 mL) was stirred at 70°C for 4 h. The mixture was concentrated under reduced pressure, diluted with EtOAc, and washed with 1 N HCI and water. The organic phase was separated, dried over MgSO4, filtered and the solvent was evaporated under reduced pressure. The compound was purified by flash chromatography on silica gel (15-40 μιτι, 120 g, heptane/EtOAc 80/20). The pure fractions were combined and evaporated to dryness to give, after crystallization from diisopropyl ether, te/t-butyl 4-(3-((1 -(4-chlorophenyl)-2-oxo-2-(6-(trifluoro-methoxy)indolin-1 -yl)ethyl)amino)-5-methoxyphenoxy)butanoate 4c (2.6 g).
Synthesis of Compound 4 and chiral separation into Enantiomers 4A and 4B: A solution of terf-butyl 4-(3-((1 -(4-chlorophenyl)-2-oxo-2-(6-(trifluoromethoxy)-indolin-1 -yl)ethyl)amino)-5-methoxyphenoxy)butanoate 4c (2.4 g, 3.8 mmol) in 4M HCI in dioxane (24 mL) was stirred at 5°C for 3 h and at room temperature for 3h. The precipitate was filtered off and dried to afford 4-(3-((1 -(4-chlorophenyl)-2-oxo-2-(6-(trifluoromethoxy)indolin-1 -yl)ethyl)amino)-5-methoxyphenoxy)butanoic acid as an HCI salt (Compound 4, 2 g, 0.8 equiv. HCI, 0.07 equiv. H2O). Compound 4 (2 g, HCI salt) was neutralized prior to chiral separation by treatment of a solution of Compound 4 (HCI salt) in ethylacetate with 1 N NaOH and evaporation of the organic layer under reduced pressure. The enantiomers were separated via Preparative Chiral SFC (Stationary phase: Chiralcel® OD-H 5 μηη 250 x 30 mm, Mobile phase: 50% CO2, 50% iPrOH (+ 0.3% iPrNH2)) and further purified via Preparative achiral SFC (Stationary phase: Cyano® 6 μιτι 150×21 .2mm, Mobile phase: 80% CO2, 20% MeOH (+ 0.3% iPrNH2)). The product fractions were combined and evaporated under reduced pressure. The two enantiomers were taken up with EtOAc and washed with 1 N HCI. The organic layers were separated, dried over MgSO4, filtered and the solvent was evaporated under reduced pressure. The first eluted enantiomer was solidified from ether/diisopropyl ether to give Enantiomer 4A (616 mg). The second eluted enantiomer was solidified from ether/diisopropyl ether to give Enantiomer 4B (715 mg).
It is also possible to separate the enantiomers starting from the HCI salt of the racemate using the same conditions for chiral separation.
Compound 4:
1H NMR (500 MHz, DMSO-c/6) δ ppm 1 .87 (quin, J=6.9 Hz, 2 H) 2.34 (t, J=7.3 Hz, 2 H) 3.07 – 3.28 (m, 2 H) 3.62 (s, 3 H) 3.85 (t, J=6.5 Hz, 2 H) 4.04 (td, J=10.5, 7.1 Hz, 1 H) 4.52 (td, J=10.3, 6.5 Hz, 1 H) 5.57 (s, 1 H) 5.76 (t, J=2.2 Hz, 1 H) 5.90 – 6.00 (m, 2 H) 7.01 (dd, J=8.2, 1 .6 Hz, 1 H) 7.33 (d, J=8.2 Hz, 1 H) 7.41 – 7.48 (m, 2 H) 7.55 (d, J=8.5 Hz, 2 H) 8.03 (s, 1 H)
LC/MS (method LC-B): Rt 2.70 min, MH+ 579
Melting point: 150°C
Enantiomer 4A:
1H NMR (500 MHz, DMSO-c/6) δ ppm 1 .87 (quin, J=6.7 Hz, 2 H) 2.34 (br t, J=7.3 Hz, 2 H) 3.08 – 3.27 (m, 2 H) 3.62 (s, 3 H) 3.85 (br t, J=6.3 Hz, 2 H) 3.99 -4.1 1 (m, 1 H) 4.47 – 4.57 (m, 1 H) 5.57 (br s, 1 H) 5.76 (s, 1 H) 5.95 (br d, J=10.1 Hz, 2 H) 6.45 (br s, 1 H) 7.01 (br d, J=7.6 Hz, 1 H) 7.34 (br d, J=7.9 Hz, 1 H) 7.44 (br d, J=8.5 Hz, 2 H) 7.55 (br d, J=8.2 Hz, 2 H) 8.04 (br s, 1 H) 12.12 (br s, 1 H) LC/MS (method LC-A): Rt 2.95 min, MH+ 579
[a]D20: -48.5° (c 0.27, DMF)
Chiral SFC (method SFC-A): Rt 1 .13 min, MH+ 579, chiral purity 100%.
Enantiomer 4B:
1H NMR (500 MHz, DMSO-c/6) δ ppm 1 .87 (br t, J=6.8 Hz, 2 H) 2.34 (br t, J=7.3 Hz, 2 H) 3.09 – 3.27 (m, 2 H) 3.62 (s, 3 H) 3.85 (br t, J=6.1 Hz, 2 H) 3.99 -4.10 (m, 1 H) 4.46 – 4.59 (m, 1 H) 5.57 (s, 1 H) 5.76 (br s, 1 H) 5.95 (br d, J=10.1 Hz, 2 H) 6.45 (br s, 1 H) 7.01 (br d, J=7.9 Hz, 1 H) 7.34 (br d, J=7.9 Hz, 1 H) 7.44 (br d, J=8.2 Hz, 2 H) 7.55 (br d, J=8.2 Hz, 2 H) 8.04 (br s, 1 H) 12.12 (br s, 1 H) LC/MS (method LC-A): Rt 2.94 min, MH+ 579
[a]D20: +42.9° (c 0.28, DMF)
Chiral SFC (method SFC-A): Rt 2.13 min, MH+ 579, chiral purity 100%.
Patent
WO2021094563
The compounds of formula I according to the present invention may be synthesized according to methods described in the art, as disclosed in WO 2016/180696. The compounds of formula II according to the present invention may be prepared according to methods described in the art, as disclosed in WO2017/167951.
Compound (b) of the present invention was tested in AG129 mouse viremia model. The synthesis of compound (b) is described in WO 2017/167951, under Example 4.
compound (b)
PATENT
WO 2018215316
The compounds of formula (I) of the present invention all have at least one asymmetric carbon atom as indicated in the figure below by the carbon atom labelled with * :
Ref
https://doi.org/10.1038/s41586-021-03990-6
////////////////////JNJ-A07, DENGUE, VIRUS, PRECLINICAL