














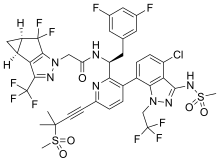
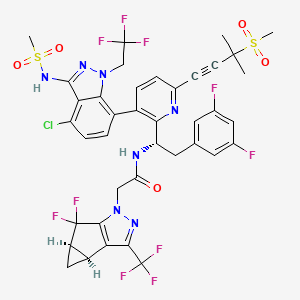
Lenacapavir sodium
レナカパビルナトリウム
| Formula |
C39H31ClF10N7O5S2. Na
C39H32ClF10N7O5S2 FREE FORM
|
|---|---|
| CAS |
2283356-12-5
2189684-44-2 FEE FORM
|
| Mol weight |
990.2641
968.28 FREE FORM
|
2022/8/17 EMA APPROVED, SUNLECA
N-[(1S)-1-[3-[4-chloro-3-(methanesulfonamido)-1-(2,2,2-trifluoroethyl)indazol-7-yl]-6-(3-methyl-3-methylsulfonylbut-1-ynyl)pyridin-2-yl]-2-(3,5-difluorophenyl)ethyl]-2-[(2S,4R)-5,5-difluoro-9-(trifluoromethyl)-7,8-diazatricyclo[4.3.0.02,4]nona-1(6),8-dien-7-yl]acetamide
|
Treatment of HIV-1 infection
|
PF-3540074, to GS-CA1, GS-6207, GS-HIV, GS-CA1, GS-CA2
Lenacapavir, sold under the brand name Sunlenca, is a medication used to treat HIV/AIDS.[1] It is taken by mouth or by subcutaneous injection.[1]
The most common side effects include reactions at the injection site and nausea.[1]
Lenacapavir was approved for medical use in the European Union in August 2022.[1]
HIV/AIDS remains an area of concern despite the introduction of numerous successful therapies, mainly due to the emergence of multidrug resistance and patient difficulty in adhering to treatment regimens.1,2 Lenacapavir is a first-in-class capsid inhibitor that demonstrates picomolar HIV-1 inhibition as a monotherapy in vitro, little to no cross-resistance with existing antiretroviral agents, and extended pharmacokinetics with subcutaneous dosing.1,2,3,5
Lenacapavir was first globally approved by the European Commission to treat adults with multi-drug resistant HIV infection.7 It is currently being investigated in clinical trials in the US.
U.S. Patent Application No. 15/680,041 discloses novel compounds useful for treating a Retroviridae viral infection, including an infection caused by the HIV virus. One specific compound identified therein is a compound of formula I:
PATENTS
- WO 2018/035359 A1
- Different formulations and salts: WO 2019/035904 A1; WO 2019/035973 A1
PATENT
WO 2019/161280 A1
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019161280
I. Synthesis of Starting Materials and Intermediates
Example la: Preparation of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan- 1-amine (VIII-02), or a co-crystal, solvate, salt, or combination thereof, and starting materials and/or intermediates therein
wherein R4 and R5 are each independently hydrogen, methyl, phenyl, benzyl, 4-nitrobenzyl, 4-chlorobenzyl, 4-brornobenzylamine, or 4-methoxybenzyl
Synthesis of 3,6-dibromopicolinaldehyde (1a)
[00553] A dry reaction flask with magnetic stir-bar was charged with 2,5-dibromopyridine (1.0 g). The flask was inerted under nitrogen, THF (4.2 mL) was added, and the thin slurry agitated. Separately, a dry glass reactor was charged with 2,2,6,6-tetramethylpiperidinylmagnesium chloride, lithium chloride complex (TMPMgCl●LiCl) (5.8 mL, 6.3 mmol). The TMPMgCl●LiCl solution was agitated and cooled to about -20 °C. The 2,5-dibromopyridine solution was added to the TMPMgCl●LiCl solution over about 30 min, maintaining a temperature below about -18 °C. Upon completing the addition, the flask was rinsed forward to the reactor with three additional portions of THF (1 mL x 2), and aged at about -20 for about 1 hour. A solution of N,N-dimethylformamide (1.6 mL, 20 mmol) in THF (1.6 mL) was added to the reactor over about 15 min. The reaction mixture was aged for a further 15 min. and quenched by the addition of a solution of acetic acid (1.9 mL, 34 mmol) in water (10 mL) over about 20 minutes, maintaining a temperature of no more than about 0 °C. To the reactor was added isopropyl acetate (10 mL) and the reaction mixture was warmed to about 20 °C. After aging for 30 min, the mixture was filtered through diatomaceous earth and the reactor rinsed with a mixture of isopropyl acetate (10 mL), saturated aqueous ammonium chloride (10 mL) and 0.2 M aqueous hydrochloric acid (10 mL). The reactor rinse was filtered and the pH of the combined reaction mixture was adjusted to about 8-9 by the addition of a 10% aqueous sodium hydroxide solution (about 6 mL). The mixture was filtered a second time to remove magnesium salts and transferred to a separatory funnel. The phases were separated and the aqueous phase was extracted with isopropyl acetate (3 x 10 mL). The combined organic extracts were washed with 50% saturated aqueous sodium chloride (20 mL), dried over anhydrous sodium sulfate, and filtered. The solution was concentrated to dryness by rotary evaporation and purified by chromatography (eluting with 0-100% ethyl acetate in heptane) to afford 3,6-dibromopicolinaldehyde (1a) as a solid. 1H NMR (400 MHz, DMSO-d6) δ 9.94 (q, J = 0.6 Hz, 1H), 8.19 (dq, J = 8.4, 0.6 Hz, 1H), 7.82 (dt, J = 8.4, 0.7 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 189.33, 148.59, 145.66, 140.17, 133.19, 120.27.
Synthesis of 3,6-dibromopicolinaldehyde (1a)
[00554] A solution of 2,5-dibromo-6-methylpyridine (8.03 g) in THF (81 mL) was cooled to about 0 °C. To this solution was charged tert-butyl nitrite (4.33 g), followed by a dropwise addition of potassium tert-butoxide (28 mL, 1.5 equiv, 20 wt% solution in THF). The reaction mixture was agitated at about 0 °C until the reaction was complete. The reaction mixture was diluted with THF (24 mL), and quenched with ammonium chloride (6.38 g, 119 mmol) in water (43 mL). The reaction mixture was distilled under vacuum to approximately 55 mL to afford a slurry, which was filtered and washed twice with water (2x 24 mL) to afford 1h. 1H NMR (400 MHz, DMSO-d6) δ 11.69 (s, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.67 (s, 1H), 7.61 (d, J = 8.5 Hz, 1H).
[00555] A solution of glyoxylic acid (407 L, 50 wt% in water) was heated to about 80 °C and in portions was charged with 1h (40.69 kg, 145.4 mol) . Reaction mixture was held at this temperature until the reaction was complete. The reaction mixture was cooled to about 20 °C, filtered, and the filter cake was washed with water until the filtrate had a pH ≥ 5, to afford 1a. 1H NMR (400 MHz, DMSO-d6) δ 9.95 (s, 1H), 8.22 (d, J = 8.4 Hz, 2H), 7.85 (d, J = 8.4 Hz, 1H).
Synthesis of (E)-N-benzhydryl-1-(3,6-dibromopyridin-2-yl)methanimine (1b-02)
[00556] Compound 1a (5.0 g, 18.0 mmol) in toluene (20 mL) was heated to about 50 °C and benzhydrylamine (3.47 g, 18.9 mmol) was charged in one portion and agitated at this temperature until the reaction was deemed complete. Methanol (61 mL) was charged and the reaction mixture was distilled to a volume of approximately 25 mL. Methanol (40 mL) was charged and the reaction mixture was distilled to a volume of approximately 30 mL. The resulting slurry was filtered and rinsed with two portions of methanol (15 mL each) and dried under vacuum to afford 1b-02.
[00557] Alternatively, compound 1a (10.0 g, 37.8 mmol) in 2-methyltetrahydrofuran (50 mL) was heated to about 50 °C and benzhydrylamine (7.28 g, 39.7 mmol) was charged dropwise. The reaction was agitated at this temperature until it was deemed complete. The reaction mixture was distilled to a volume of approximately 30 mL. To the reaction mixture was charged heptane (100 mL) and 1b-02 seed (59.3 mg, 0.138 mmol). The resulting slurry was filtered, rinsed with two portions of heptane (2x 20 mL), and dried under vacuum to afford 1b-02. 1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.44 – 7.40 (m,
4H), 7.38 – 7.32 (m, 4H), 7.28 – 7.22 (m, 2H), 5.88 (s, 1H).
Synthesis of (E)-N-benzhydryl-1-(3,6-dibromopyridin-2-yl)methanimine (1b-02)
[00558] 1a (2.00 g) was combined with isopropanol (7.6 mL) and agitated at ambient temperature. To this mixture was added potassium metabisulfite (0.96 g) in water (3.8 mL), dropwise. This mixture was agitated for at least 90 minutes and the resulting slurry was filtered. The filter cake was rinsed twice with isopropanol (6 mL then 12 mL) to afford 1i-1. 1H NMR (400 MHz, DMSO-d6) δ 7.92 (d, J = 8.3 Hz, 1H), 7.47 (d, J = 8.3 Hz, 1H), 5.48 – 5.38 (m, 2H).
[00559] li-1 (1.00 g) was combined with 2-methyltetrahydrofuran (3.5 mL) and agitated at ambient temperature. To this slurry was charged potassium hydroxide (443.8 mg, 7.91 mmol) in water (4 mL) and the biphasic mixture was agitated for 2 hours. The layers were separated and the aqueous layer was extracted with an additional portion of 2-methyltetrahydrofuran (3.5 mL). To the combined organics was charged benzhydrylamine (0.47 mL, 2.7 mmol). The reaction mixture was concentrated in vacuo (-300 mbar, 45 °C bath) to a volume of approximately 3 mL. Heptane (7 mL) was charged and the mixture was agitated. The resulting slurry was filtered to afford 1b-02 1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.44 – 7.40 (m, 4H), 7.38 – 7.32 (m, 4H), 7.28 – 7.22 (m, 2H), 5.88 (s, 1H).
Synthesis of (E)-N-benzhydryl-1-(3,6-dibromopyridin-2-yl)methanimine (1b-02)
[00560] Compound 1a (1.0 g) was added to a reactor, and toluene (6.0 mL) was added to the reactor. The mixture was agitated. Aminodiphenylmethane (0.73 g, 1.05 equiv.) was added to the reaction mixture. The jacket was heated to about 60 °C, and the mixture was allowed to age for about 1 hour. After about one hour, the mixture was carried forward to the next step. 1H NMR (400 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.05 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.4 Hz, 4H), 7.40 – 7.34 (m, 7H), 7.29 (td, J = 6.9, 6.5, 1.7 Hz, 5H), 7.22 – 7.16 (m, 3H), 5.81 (s, 1H).
Synthesis of N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-1,1-diphenylmethanimine (1d-02)
[00561] A solution of1b-02 in toluene (1.0 g in 3.8 mL) was stirred in a reactor at about 60 °C. Tetrabutylammonium bromide (0. 08 g, 0.10 equiv.) was added, 3,5-difluorobenzylbromide (0.60 g, 1.20 equiv.) was added, and potassium hydroxide (50% in water, 1.3 g, 5 equiv.) was added. The mixture was aged for about 4 hours and sampled for conversion. When the reaction was complete, the aqueous phase was removed, and water (3.1 mL) was added to the reactor. Contents were agitated and phases were allowed to settle. The aqueous phase was removed, and the toluene solution of1d-02 was carried forward to the next step. 1H NMR (400 MHz, Chloroform-d) δ 7.78 (dd, J = 8.6, 1.0 Hz, 1H), 7.64 – 7.60 (m, 2H), 7.59 – 7.53 (m, 1H), 7.49 (d, J = 8.3 Hz, 1H), 7.47 (s, 0H), 7.45 (s, 0H), 7.43 (d, J = 0.7 Hz, 0H), 7.41 – 7.34 (m, 3H), 7.33 (t, J = 1.4 Hz, 1H), 7.28 (t, J = 7.3 Hz, 2H), 7.22 (s, 0H), 7.18 (d, J = 8.3 Hz, 1H), 6.87 (dd, J = 7.7, 1.7 Hz, 2H), 6.55 (dt, J = 9.0, 2.3 Hz, 1H), 6.50 (dd, J = 7.0, 4.9 Hz, 3H), 5.26 (s, 0H), 5.16 (t, J = 6.9 Hz, 1H), 3.32 (dd, J = 13.2, 6.6 Hz, 1H), 3.16 (dd, J = 13.1, 7.2 Hz, 1H).
Synthesis of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (X) from N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-1,1-diphenylmethanimine (1d-02)
[00562] A solution of 1d-02 in toluene (1.0 g in 3.0 mL) was stirred in a reactor at about 60 °C. Sulfuric acid (0.93 g, 5 equiv.) was diluted into water (3.5 mL), and added to the reactor. The mixture was aged for about 4 hours. When the reaction was complete, the aqueous phase was removed. The aqueous phase was recharged to the reactor, and heptane (2.5 mL) was added. The mixture was agitated and agitation stopped and layers allowed to settle. The aqueous phase was removed, and heptane was discharged to waste. Toluene (5.0 mL) and potassium hydroxide (50% in water, 2.1 g, 10 equiv.) was added to the reactor. The aqueous acidic solution was added to the reactor. The mixture was agitated for about 10 minutes, and agitation stopped and phases allowed to settle. The aqueous phase was discharged to waste. Water (2.5 mL) was added to the reactor, and the mixture was agitated for about 5 minutes, and agitation was stopped and the phases were allowed to settle. The aqueous phase was discharged to waste. The toluene solution of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (X) was carried forward to the next step. 1H NMR (400 MHz, Chloroform-d) δ 7.60 (d, J = 8.3 Hz, 1H), 7.21 (d, J = 8.3 Hz, 1H), 6.74 – 6.67 (m, 2H), 6.66 – 6.58 (m, 1H), 4.57 – 4.45 (m, 1H), 3.02 (dd, J = 13.5, 5.2 Hz, 1H), 2.72 (dd, J = 13.5, 8.6 Hz, 1H), 1.77 (s, 3H).
Synthesis of (S)-1-(3.6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (R)-2-hydroxy-2-phenyl acetate (VIII-03)
[00563] A solution of X in toluene (1.0 g in 7.1 mL) was stirred in a reactor at about 60 °C. The mixture was distilled to minimum volumes (2.9 mL), and methyl tert-butyl ether was added (7.1 mL). (R)-(-)-Mandelic acid (0.41 g, 1 equiv.) was added, and the mixture was cooled to about 0 °C. The newly formed slurry was filtered, providing (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (R)-2-hydroxy-2-phenylacetate (VIII-03). 1H NMR (400 MHz, DMSO-d6) δ 7.93 (d, J = 8.4 Hz, 1H), 7.49 (d, J = 8.4 Hz, 1H), 7.34 (d, J = 7.3 Hz, 2H), 7.28 – 7.14 (m, 4H), 7.01 (tt, J = 9.4, 2.3 Hz, 1H), 6.79 (d, J = 7.4 Hz, 3H), 4.77 (s, 1H), 4.55 (d, J = 6.6 Hz, 1H), 3.02 (s, 1H), 2.92 (d, J = 6.7 Hz, 2H), 1.05 (s, 2H).
Synthesis of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine N-acetyl-D- Leucine (VIII-04)
[00564] A reactor was charged with X (15.0 g), N-acetyl-D-leucine (8.28 g) and zinc oxide (0.311 g). Toluene (375 mL) was charged to the reactor followed by 2-pyridinecarboxaldehyde (183 μL). The mixture was aged at about 55 °C for about 6 hrs. and then held at about 35 °C for about 4 days. The mixture was cooled to about 0 °C and held for about 17 hrs. The product was isolated by filtration and the filter cake was washed with cold toluene (2 x 75 mL). The filter cake was re-charged to the reactor. Ethanol (150 mL) was added and the mixture distilled to remove residual toluene. Once the toluene was removed, the reactor volume was adjusted with ethanol to about 90 mL and the mixture was cooled to about 25 °C. Water (210 mL) was added over approximately 10 min. and the mixture aged for approximately 12 hrs. The slurry was filtered and the solids were dried to afford VIII-04. 1H NMR (400 MHz, DMSO-d6) δ 8.03 (d, J = 8.0 Hz, 1H). 7.95 (d, J = 8.3 Hz, 1H), 7.49 (d, 7 8.3 Hz, 1H), 7.03 (tt, J = 9.5, 2.4 Hz, 1H),
6.87 (dtd, J = 8.4, 6.2, 2.2 Hz, 2H), 5.49 (s, 3H), 4.42 (dd, J = 7.9, 5.9 Hz, 1H), 4.18 (q, J = 7.8 Hz, 1H), 2.93 (dd, J = 13.3, 5.9 Hz, 1H), 2.85 (dd, J = 13.2, 8.0 Hz, 1H), 1.83 (s, 3H), 1.71 -1.54 (m, 1H), 1.47 (dd, J = 8.4, 6.2 Hz, 2H), 0.88 (d, J = 6.6 Hz, 3H), 0.83 (d, J = 6.5 Hz, 3H).
13C NMR (101 MHz, DMSO-d6) δ 174.72, 169.03, 162.07 (dd, J = 245.5, 13.3 Hz), 161.79, 143.51, 142.82 (t, J = 9.4 Hz), 139.72, 128.39, 119.30, 113.36 – 111.39 (m), 101.73 (t, J = 25.7 Hz), 55.19, 50.69, 41.74 (d, J = 2.3 Hz), 40.51, 24.36, 22.91, 22.44, 21.46.
Example 1b: Preparation of alternative starting materials and intermediates for use in the formation of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difliiorophenyl)ethan-1-amine (VIII), or a co-crystal, solvate, salt, or combination thereof
Synthesis of (R)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-ol (XII)
[00565] A stainless steel autoclave equipped with a glass inner tube was charged with compound XI (1.00 g) and (A)-RuCY-XylBINAP (16 mg, 0.05 equiv.). EtOH (1.0 mL) and IPA (1.0 mL) followed by tert-BuOK (1.0 M solution in THE, 0.51 mL, 0.2 equiv.) were added to the autoclave. After being purged by H2, the autoclave was charged with 3 MPa
of H2. The mixture was stirred at about 20 °C for about 10 h. To the mixture, cone. HCl aqueous solution was added and pH was adjusted to 2. 1H NMR (400 MHz, CDCl3): δ 7.72 ( d, J = 8.2 Hz, 1H), 7.33 (d, J = 8.2 Hz, 1H), 6.80 -6.72 (m, 2H), 6.68 (tt, J = 9.2, 2.4 Hz, 1H), 5.16 (dd, J = 8.2, 3.4 Hz, 1H), 3.60 (br, 1H), 3.12 (dd, J = 13.8, 3.4 Hz, 1H), 2.81 (dd, J = 13.8, 8.2 Hz,
1H). 13C NMR (100 MHz, CDC13): d 162.8 (dd, J= 246.4, 12.9 Hz), 160.1, 143.0, 141.3 (t, j = 9.1 Hz), 139.8, 128.7 (t, J= 35.7 Hz), 117.9, 112.3 (m), 102.1 (t, J= 25.0 Hz), 72.0, 43.0. 19F NMR (376 MHz, CDCl3): δ -112.1 (m).
Synthesis of N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-15-chloranimine (X-02)
[00566] Compound XIII (.0 g) was dissolved in THF (4.2 mL) and was cooled over an ice bath. Diphenylphosphoryl azide (0.66 mL, 1.2 equiv.) was added followed by DBU (0.46 mL, 1.2 equiv.) over about 25 min at below about 4 °C. The dark mixture was aged about 1 hour, and the cooling bath was removed. After about 2.5 hours age at RT, some starting material was still present so more diphenylphosphoryl azide (0.15 equiv.) and DBU (0.15 equiv.) were added after cooling over an ice bath. After about 2 hours, more diphenylphosphoryl azide (0.08 equiv.) and DBU (0.08 equiv.) were added. The reaction mixture was allowed to age overnight for about 16 h to allow the conversion to azide intermediate complete. The reaction mixture was cooled over an ice bath and triphenylphosphine (1.0 g, 1.5 equiv.) was added over about 15 min at about 6 °C). The cooling bath was removed after about 10 min and the reaction mixture was agitated for additional about 2.5 hours. To this reaction mixture was added water (0.18 mL, 4 equivalents) and the resulting mixture was aged for about 15 hours at room temperature. The mixture was diluted with EtOAc (5.0 mL) and was washed with water (4.2 mL + 2.0 mL). The aqueous layer was back extracted with EtOAc (4.0 mL) and the EtOAc layer was washed with water (1.0 mL). The organic layers were combined, concentrated via rotary evaporation and evaporated with EtOAc (4 x 4.0 mL) to dry. The residue was dissolved to a 50 ml solution in EtOAc, and cooled over an ice bath to become slurry. To the cold slurry 4N HCl/dioxane (0.76 mL, 1.2 equiv.) was added and the slurry was aged about 2 hours at room temperature. The solid product was filtered and the filter cake was rinsed with EtOAc and dried at about 35 to 50 °C under vacuum to give X-02.
[00567] Recrystallization: A portion of the above obtained X-02 (1.0 g) was mixed with EtOAc (10 mL) and was heated to 65 °C to afford thick slurry. The slurry was aged at about 65 °C for about 2 hours, and overnight at room temperature. The solids were filtered with recycling the mother liquor to help transfer the solids. The filter cake was rinsed with EtOAc, and dried overnight at about 50 °C vacuum to afford X-02. 1H NMR (300 MHz, DMSO-d) δ 8.78 (br s, 3 H), 8.06-8.02 (m, 1 H), 7.64-7.61 (m, 1 H), 7.15-7.08 (m, 1 H), 6.83-6.78 (m, 2 H), 4.87-4.82 (m, 1 H), 3.35-3.25 (m, 1 H), 3.17-3.05 (m, 1 H). 19F NMR (282.2 MHz, Chloroform-d) δ – 109.9-110.1 (m).
Synthesis of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl methanesulfonate (XIII-A)
[00568] Compound XIII (1.0 g) and DMAP (0.1 equiv.) were dissolved in THF (4.5 mL) and cooled over an ice bath. Triethylamine (Et3N) (0.39 mL, 1.1 equiv.) was added followed by methanesulfonyl chloride (218 μL, 1.1 equiv.). The cooling bath was removed, and the mixture was aged about 1.5 hours at room temperature. The reaction mixture was cooled over an ice bath and quenched with water (10 mL). The mixture was diluted with EtOAc and the phases were separated. The aqueous phase was extracted with EtOAc, and the combined organic phase was dried (Na2SO4) and was passed through silica gel with EtOAc. The filtrate was concentrated to afford the mesylate (XIII-A). 1H NMR (300 MHz, Chloroform-d) δ 7.72-7.66 (m, 1 H), 7.38-7.32 (m, 1 H), 6.78-6.63 (m, 3 H), 6.17-6.13 (m, 1 H), 3.40-3.25 (m, 2 H), 2.87 (s, 3 H). 19F NMR (282.2 MHz, Chloroform-d) δ -109.3—109.5 (m).
Synthesis of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (X) from 1-(3,6- dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl methanesulfonate (XIII-A)
[00569] A glass pressure bottle was charged with the mesylate (XIII-A) (1.0 g), 28-30% ammonium hydroxide (19 mL) and MeOH (4.7 mL). The mixture was sealed and heated at about 70 °C for about 16 hours, and extracted with 2-MeTHF/ EtOAc. The organic layer was dried (Na2SO4) and purified by silica gel chromatography (10-60% EtOAc/hexanes) to afford racemic amine X. 1H NMR (300 MHz, Chloroform-d) δ 7.70-7.60 (m, 1 H), 7.30-7.20 (m, 1 H), 6.78-6.60 (m, 3 H), 4.46-4.58 (m, 1 H), 3.00-3.16 (m, 1 H), 2.70-2.80 (m, 1 H). 19F NMR (282.2 MHz, Chloroform-d) δ -110.3 – 110.4 (m).
Synthesis of (Z)-N-(1-(3,6-dibrornopyridin-2-yl)-2-(3,5-difluorophenyl)vinyl)acetamide (1f)
[00570] A glass reactor was charged with XI (1.0 g). Ethanol (5.0 mL) was added, and the slurry was agitated while hydroxylamine hydrochloride (0.88 g) was charged. Pyridine (1.0 mL) was added and the mixture heated at about 55-65 °C for about two hours. The mixture was cooled to about 20 °C, transferred to a flask, and concentrated to approximately 75 mL by rotary evaporation. The concentrate was returned to the reactor, rinsing through with isopropyl acetate (5.0 mL). Residue remaining in the flask was carefully (gas evolution) rinsed into the reactor with saturated aqueous sodium bicarbonate (5.0 mL). The bi-phasic mixture was agitated, the phases separated, and the organic extract washed with water (3.2 mL) and saturated sodium chloride (3.2 mL). The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated to dryness by rotary evaporation to yield 1e which was used without further purification.
[00571] A glass reactor was charged with iron powder (<10 micron, 0.30 g mmol) followed by acetic acid (1.6 mL) and acetic anhydride (0.72 mL). The slurry was de-gassed by holding the reactor contents under vacuum until bubbling was observed, and back-filled with nitrogen (3 cycles). The mixture was heated at 115-120 °C for 2 hours and cooled to 40 °C. Compound le from the previous step in isopropyl acetate (2.0 mL) was added over 30 min. Upon completing the addition, the temperature was raised to 45-65 °C and the mixture aged for about 2 hours. A slurry of diatomaceous earth (1.0 g) in isopropyl acetate (2.0 mL) was added, followed by toluene (2.0 mL). The slurry was filtered, hot, through a Buchner funnel and the reactor and filter cake were washed with warm isopropyl acetate (3 x 1.8 mL). The filtrate was transferred to a reactor and the solution washed with 0.5% aqueous sodium chloride (4.2 mL). Water (3.1 mL) was added to the reactor and the mixture was cooled to about 5 °C. The pH was adjusted to 7-9 with the addition of 50 wt% aqueous sodium hydroxide; following separation, the organic extract was warmed to room temperature and washed with aqueous 1% (w/w) sodium chloride NaCl (3.6 mL). The organic extract was discharged to a flask and dried over anhydrous sodium sulfate (ca. 0.8 g), filtered through diatomaceous earth, and concentrated to approximately 4 mL at 100 mmHg and 45 °C water bath. The warm solution was returned to the reactor, rinsing forward with isopropyl acetate to a produce a total volume of approximately 5.2 mL. This solution was heated further to 50 °C with agitation, cooled to about 35 °C, and seeded with pure 1f (0.006 g). Heptane (9.6 mL) was added over a period of about 4 hours, the solution was cooled to about 10 °C, and the product was isolated by filtration. The filter cake was washed with 33.3% iPAc in heptane (4.0 mL) and dried in a vacuum oven at 40 °C with nitrogen sweep for approximately 24 hours. Compound 1f, a mixture of geometric isomers (approximately 94:6 ratio) was isolated. Major isomer: 1H NMR (400 MHz, DMSO-d6) δ 9.96 (s, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.66 (d, J= 8.4 Hz, 1H), 7.05 (s, 1H), 6.97 (tt, J = 9.2, 2.2 Hz, 1H), 6.40 – 6.31 (m,
2H), 1.97 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.37, 162.04 (dd, J = 245.1, 13.9 Hz), 154.47, 143.63, 139.45, 139.40 – 139.18 (m), 135.99, 129.44, 120.66, 113.80, 111.23 – 109.68 (m), 101.77 (t, J = 26.0 Hz), 23.49.
Synthesis of (S)-N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)acetamide (1g)
[00572] Preparation of catalyst solution: A flask was charged with [IrCl(cod)((S)-segphos)] (110 mg) and the internal atmosphere was replaced with N2. EtOAc (200 mL) was added to the flask and the mixture was stirred until the catalyst solid was dissolved.
[00573] A stainless steel autoclave was charged with compound 1f (1.0 mg). EtOAc (16 mL) and followed by the catalyst solution prepared above (4.0 mL, 0.001 equiv.) were added to the autoclave. After being purged by H2, the autoclave was charged with 3 MPa of H2.The mixture was stirred at about 130 °C for about 6 hours and cooled to room temperature and H2 was vented out. The reaction mixture was purified by silica gel column chromatography (EtOAc/Hexane = 1/4 to 1/1) to afford 1g. 1H NMR (400 MHz, CD2Cl2): d 7.70 ( d, J = 8.0 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 6.68 (tt, J = 9.2, 2.4 Hz, 1H), 6.64 -6.58 (m, 2H), 6.49 (brd, j = 8.0 Hz, 1H), 5.74 (ddt, J = 8.0, 7.2, 6.4 Hz, 1H), 3.10 (dd, J = 13.6, 6.4 Hz, 1H), 2.99 (dd, J = 13.6, 7.2 Hz), 1.95 (s, 3H). 13C NMR (100 MHz, CD2Cl2): δ 169.5, 163.3 (dd, J = 246.0, 12.9 Hz), 159.1, 143.6, 141.4 (t, J = 9.1 Hz), 140.7, 129.1, 119.9, 112.9 (m), 102.6 (t, J= 25.1 Hz), 53.0, 41.3, 23.6. 19F NMR (376 MHz, CD2Cl2): δ -111.3 (m).
Synthesis of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VIII) from 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-one (XI), Method 1
[00574] A glass-lined reactor was charged with isopropylamine (about 18 g) and triethanolamine (3.8 g). Water (231 mL) was added and the pH was adjusted to about 7.5 by the addition of concentrated hydrochloric acid. A portion of the buffer solution (23 mL) was removed. The transaminase enzyme (2.5 g) was added to the reactor as a suspension in buffer solution (12 mL), followed by addition of pyridoxal phosphate monohydrate (50 mg) as a solution in buffer solution (12 mL). A solution of XI (1.0 g) in dim ethyl sulfoxide (23 mL) was added to the reactor and the mixture was heated at about 35 °C for about 48 hours with constant nitrogen sparging of the solution. The reaction mixture was cooled to about 20 °C the unpurified amine was removed by filtration. The filter cake was washed with water (3 x 7.7 mL) and the product was dried at about 60 °C under vacuum with nitrogen sweep to afford VIII.
Synthesis of (S)-1-(3.6-dibromopyridin-2-yl)-2-(3.5-difluorophenyl)ethan-1-amine (VIII) from 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-one (XI), Method 2
[00575] A stainless steel reactor was charged with XI (1.0 g) and p-toluenesulfonic acid (0.49 g). Ammonia (7 M in methanol, 3.7 mL) was added and the vessel was sealed and heated at about 60 °C for about 18 hours. The mixture was cooled to about 20 °C and sparged for about 30 min to remove excess ammonia. A solution of diacetato[(R)-5,5′-bis(diphenylphosphino)-4,4′-bi-1,3-benzodioxole]ruthenium(II) (0.10 g) in methanol (0.5 mL) was added to the reactor, which was sealed and heated at about 60 °C under a hydrogen atmosphere (400 psi) for a further about 6-10 hours. Upon cooling to about 20 °C the mixture was filtered through a plug of silica, rinsing with additional methanol (5.0 mL). Concentration of the filtrate by rotary evaporation affords VIII.
Example 1c: Preparation of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyI)ethan-1-amine (X) by racemization of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VIII)
[00576] A vial was charged with zinc acetate (25 mol %), enantioenriched VIII (1.0 g, 92:8 enantiomer ratio), toluene (10 mL), and 2-formylpyridine (5 mol %). The vial was wanned to about 60 °C and stirred for about 4 h.
Example 2: Preparation of (S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VI)
[00577] A glass-lined reactor was charged with (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (R)-mandelic acid salt (VIII-03) (1.0 g), 3-methyl-3-(methylsulfonyl)but-1-yne (IX) (about 0.3 g), and dichlorobis(triphenylphosphine)palladium(II) (about 0.39 g). The reactor was evacuated and purged with nitrogen to inert. To this reactor was added 2-methyltetrahydrofuran (6.4 kg) and triethylamine (0.92 kg 5.0 equiv.). The reaction mixture was agitated at about 65-75 °C until the reaction was deemed complete by HPLC analysis. Upon cooling to about 30-40 °C the reaction mixture was discharged to another reactor and the parent reactor was rinsed with 2-methyltetrahydrofuran (4.6 g) and the resulting solution transferred to the receiving reactor. To the reactor was added water (5.0 g) and the biphasic mixture agitated at about 30-40 °C for about 30 min. Agitation was ceased and the mixture was allowed to layer for 30 min. The lower aqueous layer was discharged and the remaining organic solution held for about 15 hours. A solution of A-acetyl-L-cysteine (196 g) and sodium hydroxide (0.80 g) in water (11.8 g) was prepared. To the reactor was added approximately half of the N-acetyl-L-cysteine solution (6.7 g). The mixture was agitated at about 55-65 °C for about 30 min. The temperature was adjusted to about 30-40 °C and agitation was ceased. After about 30 min had elapsed, the lower aqueous phase was discharged. The remaining alkaline N-acetyl-L-cysteine solution (5.4 kg) was added and the mixture was heated, with agitation, to about 55-65 °C and held for about 30 min. The temperature was adjusted to about 30-40 °C and agitation was ceased. After about 30 min had elapsed, the lower aqueous phase was discharged. To the reactor was added a solution of sodium chloride (0.26 g) in water (4.9 g) and the mixture agitated at about 30-40 °C for about 30 min. Agitation was ceased and the biphasic mixture allowed to layer for about 30 min. The lower aqueous layer was discharged and the contents cooled to about 15-25 °C and held for about 16 hours. The mixture was concentrated at about 55-65 °C. The concentrated solution was cooled to about 30-40 °C and heptane (3.4 kg) was added over about 2 hours. The resulting slurry was cooled to about 20 °C and aged for about 20 h, and filtered. The filter cake was washed with 2-methyltetrahydrofuran/heptane (1:1 v/v,2 mL) and the solids dried in a vacuum oven at about 40 °C to yield (S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VI)). 1H NMR (400 MHz, DMSO-d6) δ 8.05 (d, J = 8.2 Hz, 1H), 7.42 (d, J = 8.2 Hz, 1H), 7.01 (tt, J = 9.5, 2.4 Hz, 1H), 6.97 – 6.84 (m, 2H), 4.41 (dd, J = 8.5, 5.2 Hz, 1H), 3.20 (s, 3H), 2.93 (dd, J = 13.3, 5.2 Hz, 1H), 2.79 (dd, J = 13.3, 8.5 Hz, 1H), 1.99 (s, 2H), 1.68 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 162.25, 162.00 (dd, J = 245.2, 13.4 Hz), 143.88 (t, J= 9.4 Hz), 141.09, 139.72, 127.51, 120.08, 112.58 – 112.12 (m), 101.45 (t, J= 25.7 Hz), 87.94, 84.25, 57.24, 55.90, 42.57, 34.99, 22.19.
Example 2a: Preparation of 3-methyl-3-(methylsulfonyl)but-1-yne (IX)
[00578] Sodium methansulfmate (418.1 g), copper (II) acetate (26.6 g), N,N,N’,N’- Tetramethylethylenediamine (TMEDA, 34.0 g), and isopropyl acetate (2100 mL) were added to a reactor and the suspension was agitated at 20 – 25 °C. 3-Chloro-3-methylbut-1-yne (3-CMB,
300 g) was added slowly to maintain a constant temperature of about 20 – 25 °C. The reaction mixture was then heated to about 30 °C until the reaction was complete. The mixture was cooled to about 20 °C and washed twice with 5% aqueous sulfuric acid (600 mL). The combined
aqueous layers were then extracted with isopropyl acetate (600 mL). The combined organic layers were then washed with water (600 mL). The product was then isolated by crystallization from isopropyl acetate (900 mL) and n-heptane (1.8 kg) at about 0 °C. The wet cake was then washed with cold n-heptane to afford IX. 1H NMR (400 MHz, DMSO-d6) δ 3.61 (s, 1H), 3.07 (s, 3H), 1.55 (s, 6H); 13C NMR (10Q MHz, DMSO) d 82.59, 77.76, 56.95, 34.95, 22.77.
Example 3a: Preparation of (3bS,4aR)-3-(trifluoromethyI)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV) from lithium (Z)-2,2,2-trifluoro-1-(3-oxobicyclo[3.1.0]hexan-2-ylidene)ethan-1-olate (3a)
Synthesis of 3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazole (3b)
[00579] A reactor was charged with 3a (1.0 g) and AcOH (4.2 ml) and the resulting solution was adjusted to about 20 °C. Hydrazine hydrate (0.29 g, 1.4 equiv.) was added over about 60 min at about 17-25 °C and the reaction mixture was stirred for about 2 hours at about 20-25 °C, warmed up to about 45 to 50 °C over about 30 min, and aged at about 50 °C overnight. Water was slowly (5 mL) added at about 50 °C and product started to crystallize after addition of 5 mL of water. Another 5 mL of water was added at about 50 °C, and the slurry was cooled down to about 20 °C in about one hour and held overnight at about 20 °C. The solids were filtered, washed with water (4X 3 mL), and dried under vacuum at about 30 °C to yield 3b. 1H NMR (400 MHz, Chloroform-d) δ 2.99 (dd, J = 17.0, 6.1 Hz, 1H), 2.89 – 2.78 (m, 1H), 2.14 (dddd, J = 9.1, 7.9, 3.6, 2.5 Hz, 2H), 1.13 (td, J = 7.8, 5.1 Hz, 1H), 0.36 – 0.26 (m, 1H).
Isolation of (3bS,4aS)-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazole (3c)
[00580] Chiral purification of 3b (1.0 g) was achieved using a 8×50 mm simulated moving bed (SMB) chromatography system and Chiralpak IG (20 μ particle size) stationary phase using acetonitrile as a mobile phase to afford 3c. 1H NMR (400 MHz, Chloroform-d) δ 3.00 (dd, J = 17.0, 5.7 Hz, 1H), 2.90 – 2.77 (m, 1H), 2.21 – 2.05 (m, 2H), 1.13 (td, J = 7.8, 5.1 Hz, 1H), 0.35 – 0.27 (m, 1H).
Synthesis of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV)
[00581] A reactor was charged with water (7 mL) and CuCl2 ● 2H2O (0.09 g, 0.1 equiv). To the reactor was added pyridine (0.42 g, 1 equiv.) and 3c. tert-Butylhydroperoxide (70% in water, 5.5 g, 8 equiv.) was added over about 0.5 hour. The reaction mixture was stirred at about 20 °C for about 2.5 days and quenched with aqueous sodium metabisulfite solution (0.73 g in 2.5 mL water). The quenched reaction mixture was extracted with isopropyl acetate (20 mL), and the aqueous layer was back extracted with isopropyl acetate (2.0 ml). The organic layers were combined and washed with aqueous ethylenediaminetetraacetic acid (EDTA) solution 0.16 g EDTA 10 ml in water), the aqueous layer was dropped, and the organic layer was further washed with aqueous EDTA solution (0.015 g EDTA in 20 ml water). The washed organic layer was concentrated to dryness. To the residue was added isopropyl acetate (2.0 ml) and heptane (2.0 mL). The solution was seeded and stirred overnight at about 20 °C, further diluted with heptane (2.0 mL), and the mixture was concentrated to dryness. The residue was suspended in heptane (4.0 mL) at about 40 °C. The solid was filtered and the filter cake was washed with heptane (1.0 mL) and dried at about 40 °C to yield XV. 1H NMR (400 MHz, Chloroform-d) δ 2.84 (dt, J = 6.8, 4.2 Hz, 1H), 2.71 – 2.64 (m, 1H), 1.79 – 1.67 (m, 2H).
Example 3b: Preparation of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV) from lithium (Z)-1-((1S,5R)-4,4- dimethoxy-3-oxobicyclo[3.1.0]hexan-2-ylidene)-2,2,2-trifluoroethan-1-olate (3d-02)
[00582] Hydrazine sulfate (0.45 g, 0.95 equiv.) and ketal lithium salt 3d-02 (1.0 g) were dissolved in ethylene glycol (9.5 mL), and the solution was heated to about 40 °C for about 16 hours. Reaction was cooled to room temperature and water (9.0 mL) was added. Reaction was polish filtered andThe filtrate was collected and to this receiving flask was added water (10 mL, 2x). Slurry was cooled in ice water bath for about five hours, and filtered. Solids were washed with ice water (10 mL, 2x), deliquored, and dried to afford XV. 1H NMR (400 MHz, CDCl3) δ 11.83 (bs, 1H), 2.93 – 2.77 (m, 1H), 2.77 – 2.58 (m, 1H), 1.86 – 1.57 (m, 2H). 19F NMR (376 MHz, CDCl3) δ -61.69. 13C NMR (101 MHz, CDCl3) δ 188.56, 144.08, 142.92, 121.82, 119.15, 36.28, 31.87, 14.15.
Example 3c: Preparation of (3bS,4aR)-3-(trifiuoromethyl)-1,3b,4,4a-tetrahydro-5H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV) from (1S,2S)-2-iodo-N-methoxy-N- methylcyclopropane-1-carboxamide (3f) and 1-(4-methoxybenzyl)-4-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)-3-(trifluoromethyl)-1H-pyrazole (3i) and preparation of starting materials and/or intermediates therein
Synthesis of (1S,2S)-2-iodo-N-methoxy-N-methylcyclopropane-1-carboxamide (3f)
[00583] Starting material iodoacid 3e is a mixture of 3e and cyclopropane carboxylic acid (des-iodo 3e) with mole ratio of 3e to des-iodo 3e of 2:1 by NMR. A mixture of 3e (1.0 g),
N,O-dimethyl hydroxyl amine-HCl (0.46 g) and carbonyl diimidazole (1.72 g) in THF was stirred overnight at room temperature. The reaction mixture was diluted with water, extracted with CH2Cl2, and concentrated to afford unpurified 3f (1.8 g). The unpurified 3f was purified by column chromatography to afford 3f which was a mixture of Wei nr eb amide 3f and des-iodo-3f (about 80:20 by HPLC).
Synthesis of 1-(4-methoxybenzyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3- (trifluoromethyl)-1H-pyrazole (3i)
[00584] To a suspension of NaH (60%, 0.31 g, 1.1 equiv.) in DMF (7.5 mL), a solution of 3g (1.0 g) in DMF (7.5 mL) was added dropwise over about 15 min at about 3 to 7 °C. The reaction mixture was stirred at room temperature for about 1 h and a solution of PMBCl (1.2 g, 1.05 equiv.) in DMF (4.2 mL) was added dropwise in about 25 min at room temperature. The reaction mixture was stirred at room temperature overnight, poured into water (17 mL), and extracted with diethyl ether (3×17 mL). The ether layers were combined and washed with water (2 x 17 mL) and brine (17 mL), dried over Na2SO4, and concentrated in vacuo to give unpurified 3h. Unpurified 3h was absorbed in silica gel (4.3 g) and purified by silica gel chromatography (eluting with 5-25% EtOAc in hexanes) to give 3h (1.5 g).
[00585] To solution of iodopyrazole 3h (1.0 g) in THF (8 mL) i-PrMgCl (2M in ether, 1.8 mL, 1.1 equiv.) was added dropwise over about 10 min at below about 5 °C. The resulting solution was stirred at about 0 °C for about 70 min and 2-methoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (970 mg, 1.81 equiv.) was added at below about 6 °C. The reaction mixture was warmed up to room temperature, quenched by addition of saturated NH4Cl (20 mL), and
extracted with EtOAc (2 x 20 mL). The combined organic layer was washed with saturated NH4Cl (10 mL) and concentrated to unpurified oil, which was combined with the unpurified oil from a previous batch (prepared using 1.1 g of 3h), absorbed on silica gel (6 g), and purified via silica gel chromatography (eluting with 5-40% EtOAc/Hexanes,). Boronate 3i was obtained. 1H NMR (300 MHz, Chloroform-d) δ 7.60 (s, 1 H), 7.23-7.19 (m, 2 H), 6.90-6.85 (m, 2 H), 5.25
(s, 2 H), 3.81 (m, 3 H), 1.29 (s, 12 H).
Synthesis of (1R,2S)-N-methoxy-2-(1-(4-methoxybenzyl)-3-(trifluoromethyl)-1H-pyrazol-4-yl)-N-methylcyclopropane-1-carboxamide (3j)
[00586] A mixture of unpurified iodide 3f (1.0 g), boronate 3i (about 2.2 g), CsF (4.5 equiv.), Pd(OAc)2 (0.1 equiv.), and PPh3 (0.5 equiv.) in DMF (58 mL) was degassed by bubbling N2 and heated at about 87 °C for about 15 hours. The reaction mixture was diluted with water,
extracted with MTBE, concentrated and the unpurified product was purified by column chromatography to give 3j. 1H NMR (300 MHz, Chloroform-d) δ 7.18-7. 14 (m, 3 H), 6.86-6.82 (m, 2 H), 5.24-5.08 (m, 2 H), 3.77 (s, 3 H), 3.63 (s, 3 H), 3.05 (s, 3 H), 2.37-2.32 (m, 1 H), 1.50-1.42 (m, 1 H), 1.32-1.21 (m, 2 H).
Synthesis of (3bS,4aR)-1-(4-methoxybenzyl)-3-ftrifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta91,2-c]pyrazol-5-one (3k)
[00587] Compound 3j (1.0 g) was treated with freshly prepared LDA (3.3 eq then 0.7 equiv.) at about -67 °C for about 2.5 hours. The reaction mixture was quenched with saturated NH4Cl (12.5 mL) and diluted with MTBE (63 mL). The organic layer was washed with brine, concentrated, and purified by column chromatography to give 3k. 1H NMR (300 MHz, Chloroform-d) δ 7.36-7.33 (m, 2 H), 6.86-6.83 (m, 2 H), 5.28 (s, 2 H), 3.78 (s, 3 H), 2.73-2.65
(m, 1 H), 2.60-2.53 (1 H), 1.70-1.61 (m, 2 H).
Synthesis of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1.2-c]pyrazol-5-one (XV)
[00588] A mixture of 3k (1.0 g) and TFA (5 mL) was heated at about 75 °C for about 3 hours and concentrated. The residue was dissolved in DCM (50 mL), washed with saturated NaHCO3 and brine, concentrated, and purified by column chromatography to give XV. 1H NMR (300 MHz, Chloroform-d) δ 2.86-2.80 (m, 1 H), 2.68-2.63 (m, 1 H), 1.77-1.65 (m, 2 H).
Example 3d: Resolution of 2-(2,2,2-trifluoroacetyl)bicyclo[3.1.0]hexan-3-one (3I) with quinine
[00589] A flask was charged with 3I (1.0 g), acetone (2.5 ml), and quinine (1.7 g, 0.65 equiv). The mixture was stirred at about 15 to 25 °C for about 18 hours and the solids were isolated by filtration and washed with acetone to provide the quinine salt 3n.
Example 4a: Preparation of ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV) from (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV)
[00590] Acetonitrile (5 vol.) was added to a reactor containing XV (1.0 g). N,N-Diisopropylethylamine (0.80 g, 1.25equiv.) was added at about 0 °C. Ethyl bromoacetate (0.91 g, 1.1 equiv.) was added over about 1 hour at about 0 °C. The reaction was stirred at about 5 °C for about 30 minutes and warmed to about 10 °C. The reaction was stirred until complete as determined by HPLC, warmed to about 20 °C, and extracted with MTBE (2 vol.) and saturated NaCl (6 vol.). The aqueous layer was removed and the organic phase was concentrated and diluted with EtOH (3 vol.). The reaction was crystallized by the addition of H2O (7.8 vol.) at about 20 °C. The mixture was cooled to about 5 °C over about 2 hours and maintained at about 5 °C for about 0.5 hour. The mixture was filtered at about 5 °C and washed with cold water (4 vol). The product was dried at about 40 °C under vacuum to give XIV. 1H NMR (400 MHz, Chloroform-d) δ 4.97 (s, 2H), 4.31 – 4.17 (m, 2H), 2.77 (dddd, J= 6.4, 5.2, 2.9, 2.3Hz, 1H), 2.65 – 2.55 (m, 1H), 1.74 – 1.64 (m, 2H), 1.34 – 1.19 (m, 5H), 0.94 – 0.84 (m, 1H).
Example 4b: Preparation of ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV) from (1R,5S)-bicyclo[3.1.0]hexan-2-one (4a)
Synthesis of (1R,5R)-2,2-dimethoxybicyclo[3.1.0]hexan-3-ol (4b-02)
[00591] Potassium hydroxide (KOH) (2.2 g, 3.50 equiv.) and anhydrous methanol (13 mL) were added to a reactor and the reaction mixture was warmed to about 55 °C and agitated until
KOH solids were dissolved completely. The mixture was adjusted to about 0 to 6 °C and compound 4a (1.0 g) was slowly added while maintaining the internal temperature at NMT 6 °C. The reaction mixture was agitated for about 45 min at about 0 to 6 °C. Diacetoxy iodobenzene (PhI(OAc)2, 5.0 g, 1.5 equiv.) was added over about 2 hours while maintaining the internal temperature at NMT 6 °C. The reaction mixture was agitated for NLT 1 hour at about 0 to 6 °C. Water (10 g) and heptane (10 mL) were added to the reaction mixture and the biphasic was agitated for NLT 30 min at about 19 to 25 °C The aqueous layer was separated and washed with heptane (10 mL). The combined organic layer was extracted twice with aqueous solution of methanol (MeOH, 10 mL) and water (5 g). The combined aqueous layer was concentrated under vacuum. The aqueous layer was extracted twice with DCM (15 mL and 5 mL). The combined organic layer was concentrated and dried under vacuum. The unpurified compound 4b-02 was obtained. 1H NMR (600 MHz, CDCl3): d 3.98 (d, 1H), 3.45 (s, 3H), 3.25 (s, 3H),
2.40 (s, 1H), 2.21 (m, 1H), 1.78 (d, 1H), 1.48 (m, 1H), 1.38 (m, 1H), 0.83 (q, 1H), 0.58 (m, 1H).
13C NMR (150 MHz, CDCl3): δ 110.91, 72.19, 51.18, 49.02, 34.08, 21.66, 14.75, 8.37.
Synthesis of (1R,5R)-2,2-dimethoxybicyclo[3.1.0]hexan-3-one (4c-02)
[00592] Oxalyl chloride (0.96 g, 1.20 equiv.) and dichloromethane (10 mL) were added to a reactor and the mixture was cooled to about -78 °C. Dimethyl sulfoxide (DMSO, 1.2 g, 2.4 equiv.) was added over about 30 min while maintaining the internal temperature below about -60 °C. After agitation for about 5 min, the solution of compound 4b-02 (1.0 g) in dichloromethane (6 mL) was added over about 30 min while maintaining the internal temperature below about -60 °C and the reaction mixture was agitated for about 20 min at about -60 °C. Triethylamine (TEA, 3.1 g, 4.8 equiv.) was added over about 40 min at about -60 °C, and the reaction mixture was warmed to about 10 to 20 °C. Water (15 g) was added and the biphasic was agitated about 30 min at about 10 to 20 °C. After phase separation, the aqueous layer was back-extracted with dichloromethane (10 mL). Combined organic layer was concentrated until no distillate was observed. To the residue was added MTBE (1 mL), filtered and evaporated to afford unpurified compound 4c-02. 1H NMR (600 MHz, CDCl3): d 3.45 (s,
3H), 3.27 (s, 3H), 2.79 (ddd, 1H), 2.30 (d, 1H), 1.73 (td, 1H), 1.63 (m, 1H), 0.96 (m, 1H), 0.25 (td, 1H). 13C NMR (150 MHz, CDCl3): δ 207.75, 102.13, 50.93, 50.50, 38.87, 19.15, 9.30, 8.56.
Synthesis of lithium (Z)-1-((1S,5R)-4,4-dimethoxy-3-oxobicyclo[3.1.0]hexan-2-ylidene)-2,2,2-trifluoroethan-1-olate (3d-02)
[00593] A reactor was charged with compound 4c-02 (1.0 g), ethyl trifluoroacetate (CF3COOEt, 0.91 g, 1.0 equiv.) and tetrahydrofuran (THF, 0.5 mL) and the reaction mixture was cooled to about -10 to 0 °C. The 1M solution of lithium bis(trimethylsilyl)amide (LiHMDS, 7.0 mL, 1.10 equiv.) was added over about 40 min while maintaining the internal temperature below about 0 °C. The reaction mixture was agitated for about 2 hours at about -10 to 0 °C until the reaction was complete. After then, the reaction mixture was wanned to about 20 °C followed by charging tert-butyl methyl ether (MTBE, 10 mL) and water (10 g). After agitating for about 30 min, the organic layer was separated and the aqueous layer was back-extracted twice with mixture of MTBE (6 mL) and THF (4 mL). The combi ned organic layer was concentrated until no distillate was observed. To the unpurified solids, THF (3 mL) and heptane (15 mL) were added at about 20 °C, and the reaction mixture was cooled to about 0 °C and agitated about 1 hour. The resulting slurry was filtered and wet cake was washed with heptane (7 g) and dried under vacuum at about 40 °C to afford compound 3d-02. 1H NMR (600
MHz, DMSO-d6): d 3.31 (s, 3H), 3.27 (s, 3H) 2.01 (m, 1H), 1.42 (td, 1H), 0.96 (m, 1H), 0.08 (q, 1H). (600 MHz, CDCl3 with THF) δ 3.44 (s, 3H), 3.24 (s, 3H), 2.26 (m, 1H), 1.48 (m, 1H), 1.04 (q, 1H), 0.25 (m, 1H). 13C NMR (150 MHz, DMSO-d6): 193.20, 120.78, 118.86, 105.53,
104.04, 50.66, 49.86, 17.34, 16.20, 13.78.
Synthesis of ethyl 2-((3bS.4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
[00594] Compound 3d-02 (1.0 g), ethyl hydrazinoacetate hydrochloride (EHA-HCl, 0.60 g,
1.0 equiv.) and absolute ethanol (EtOH, 15 mL) were added to a reactor and the reaction mixture was cooled to about 0 – 5 °C. Sulfuric acid (H2SO4, 0.19 g, 0.50 equiv.) was added while maintaining the internal temperature below about 5 °C. Triethyl orthoformate (0.86 g, 1.50 equiv.) was added and the reaction mixture was agitated at about 0 to 5 °C for about 15 hours. The reaction mixture was warmed to about 20 to 25 °C and water (30 g) was added over about 15 minutes. The content was cooled to about 0 to 5 °C and agitated for about 1 hour. The slurry was filtered and wet cake was washed with water (5 g) and dried under vacuum at about 45 °C to afford XIV 1H NMR (600 MHz, CDCl3): d 4.97 (s, 1H), 4.23 (qd, 2H), 2.77 (quint. 1H), 2.60 (quint, 1H), 1.69 (m, 2H), 1.28 (t, 3H). 13C NMR (150 MHz, CDCl3): d 187.14, 165.98, 143.35, 143.12, 121.37, 119.59, 62.34, 51.83, 35.35, 31.72, 14.00, 13.73.
Example 4c: Kinetic resolution of ethyl 2-(5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro- 1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XVII) to form ethyl 2- ((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
[00595] Compound XVII (1.0 g), (R)-2-methyl-CBS-oxazaborolidine (0.0.05 g, 0.05 equiv.), and tetrahydrofuran (11.9 g) were combined and cooled to about 0 to 5 °C. A solution of borane dimethyl sulfide complex (0.14 g, 0.55 equiv.) in tetrahydrofuran (0.67 g) was added to the mixture, and the mixture was agitated at about 0 to 5 °C until the reaction was deemed complete. Methanol (1 mL) was added to the mixture at about 0 to 5 °C over about 1 h, and the mixture was adjusted to about 15 to 25 °C. The mixture was concentrated under vacuum and combined with tetrahydrofuran (2.7 g). The mixture was combined with 4-dimethylaminopyridine (0.18, 0.44 equiv.) and succinic anhydride (0.30 g, 0.87 equiv.) and agitated at about 15 to 25 °C until the reaction was deemed complete. The mixture was combined with tert-butyl methyl ether (5.2 g) and washed with 1 M aqueous HCl (6.7 g), twice with 5 wt % aqueous potassium carbonate (6.7 g each), and 5 wt % aq. sodium chloride (6.7 g). The organics were concentrated under reduced pressure to an oil which was dissolved in dichloromethane (0.1 g) and purified by flash column chromatography (2.0 g silica gel, 20:80 to 80:20 gradient of ethyl acetate:hexanes). The combined fractions were concentrated under vacuum to give XIV.
Example 4d: Preparation of (1R,5S)-bicyclo[3.1.0]hexan-2-one (4a)
[00596] 4-Tosyloxycyclohexanone (50 mg), (8α,9S)-6′-methoxycinchonan-9-amine trihydrochloride (16 mg), trifluoroacetic acid (28 μL), lithium acetate (49 mg), water (3.4 μL), and 2-methyltetrahydrofuran (0.75 mL) were combined in a vial. The mixture was agitated at about 20 °C until the reaction was complete. 4a was isolated by vacuum distillation. 1H NMR (400 MHz, CDCl3) δ2.05 (m, 5H), 1.74 (m, 1H), 1.18 (m, 1H), 0.91 (m, 1H).
Example 5: Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a- dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane]-1(3bH)- yl)acetate (5h) from (1R,5R)-2,2-dimethoxybicyclo[3.1.0]hexan-3-ol (4b-02)
Synthesis of (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan1-3-ol (5d)
[00597] A mixture of ketal alcohol 4b-02 (1.0 g), ethanedi thiol (0.91 g), MeCN (7.5 ml) and BiCl3 (0.30 g) was agitated at r.t. overnight. The solids were removed by filtration and the filtrate was concentrated and the residue was further purified by flash column on silica gel to obtain the two isomers. Major product: 1H NMR (400 MHz, Chloroform-d) δ 3.82 (ddt, J = 6.1, 1.3, 0.6 Hz, 1H), 3.41 – 3.32 (m, 2H), 3.31 -3.23 (m, 1H), 3.14 – 3.06 (m, 1H), 2.71 (s, 1H),
2.33 (dddd, J = 14.0, 6.2, 4.8, 1.4 Hz, 1H), 2.00 (d, J = 13.9 Hz, 1H), 1.79 – 1.72 (m, 1H), 1.54 -1.46 (m, 1H), 1.04 (dt, J = 5.1, 3.9 Hz, 1H), 0.63 – 0.54 (m, 1H). Minor product: 1H NMR (400 MHz, Chloroform-d) δ 3.83 (q, J = 9.1 Hz, 1H), 3.43 – 3.34 (m, 2H), 3.33 – 3.25 (m, 2H), 2.35 (d, J= 11.2 Hz, 1H), 2.18 (ddd, J = 12.7, 6.7, 0.4 Hz, 1H), 1.84 (ddd, J= 8.1, 6.3, 3.7 Hz, 1H),
1.60 – 1.51 (m, 1H), 1.43 – 1.35 (m, 1H), 0.65 (tdt, J= 8.1, 5.9, 0.8 Hz, 1H), 0.57 (dddd, J= 5.9, 4.2, 3.7, 0.6 Hz, 1H).
Synthesis of (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan1-3-one (5e)
[00598] To a dried flask was sequentially added dithiolane alcohol 5d (1.0 g), CH2Cl2 (25 ml), anhydrous DMSO (8.5 ml), and tri ethylamine (3.5 ml) and the resulting mixture was aged at room temperature for about 21 hours. The reaction mixture was transferred to a separatory funnel, diluted with CH2Cl2 (30 ml), washed with 1 M HCl (25 ml), and water (25 ml). The CH2Cl2 layer was concentrated to a solid and further purify by flash column chromatography on silica gel eluted with gradient EtOAc/n-heptane (0-20%) to obtain 5e. 1H NMR (400 MHz, Chloroform-d) δ 3.57 (dddd, J = 10.5, 5.6, 4.3, 0.5 Hz, 1H), 3.49 – 3.41 (m, 1H), 3.39 – 3.28 (m, 2H), 3.10 (ddd, J = 18.3, 5.6, 2.2 Hz, 1H), 2.29 (d, J = 18.3 Hz, 1H), 1.89 (ddd, J = 8.0, 7.0, 3.9
Hz, 1H), 1.63 (tdd, J= 7.3, 5.6, 4.1 Hz, 1H), 1.05 (tdd, J = 8.0, 6.3, 2.2 Hz, 1H), 0.21 (dt J = 6.4, 4.0 Hz, 1H).
Synthesis of lithium (Z)-2,2,2-trifluoro-1-((1R,5S)-3-oxospiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan]-4-ylidene)ethan-1-olate (5f)
[00599] To a flask with dithiolane ketone 5e (1.0 g) under N2 was added anhydrous THF (8.8 ml), and the mixture was cooled to about -78 °C and followed by addition of LiHMDS (1 M in THF, 7.4 ml) over about 5 min. The resulting mixture was agitated at about -78 °C for about 0.5 hours, and ethyl trifluoroacetate (0.88 ml) was added. The resulting mixture was agitated at about -78 °C for about 10 minutes, at about 0 °C for about 1 hour, and at room temperature overnight. THF was removed under reduced pressure and the residue was crystallized in n-heptane (about 18 ml). The solid product was isolated by filtration, and the filter cake was rinsed with n-heptane (4.1 ml), and dried at about 50 °C under vacuum to provide 5f. 1H NMR (400 MHz, Acetonitrile-d3) δ 6.98 (s, 0H), 5.20 (s, 0H), 3.60 – 3.50 (m, 2H), 3.46 – 3.36 (m, 2H), 2.28 – 2.20 (m, 1H), 1.80 (ddd, J = 8.3, 7.2, 4.1 Hz, 1H), 1.39 (s, 1H), 1.03 (ddd, J = 8.3, 6.7, 4.8 Hz, 1H), 0.17 (ddd, J = 4.7, 4.2, 3.6 Hz, 1H).
Synthesis of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydrospiro[cvciopropa[3.4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane] (5g)
[00600] To flask containing the dithiolane lithium salt 5f (1.0 g) was added water (10 ml), hydrazine hydrate (0.88 ml) and acetic acid (10 ml). The reaction mixture was heated at about 35 °C for about 2 hours, and at about 55 °C for about 2 hours. Water was removed under reduced pressure and the residue was diluted with acetic acid (20 ml) and heated at about 55 °C for about 0.5 hour and held at room temperature overnight. The reaction mixture was further heated at about 65 °C for about 20 hours, and cooled down and concentrated to remove volatile components by rotavap. The residue was triturated with water (50 ml) at about 0 °C and the solid residue was isolated and further washed with ice-cold water (2×10 ml). The solids were further dried to afford unpurified 5g. 1H NMR (400 MHz, Chloroform-d) δ 3.65 – 3.46 (m, 4H), 2.60 (dddd, J = 8.3, 5.6, 4.2, 0.7 Hz, 1H), 2.47 – 2.38 (m, 1H), 1.33 (dddd, J= 8.2, 7.4, 5.7, 0.7 Hz, 1H), 0.66 (dddd, J = 5.7, 4.3, 3.6, 0.7 Hz, 1H)
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5.2′-[1,3]dithiolane]-1(3bH)-yl)acetate
(5h) from (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane] (5g)
[00601] A reactor was charged with dithiolane pyrazole 5g (1.0 g) and THF (15 ml). The contents were adjusted to about 0 to -5 °C and followed by addition of ethyl bromoacetate (0.44 ml, 1.1 equiv.). To the resulting mixture NaHMDS (2 M, 2.0 ml, 1.1 equiv.) was added over about 10 min via syringe pump at about -2.5 to 0 °C and the mixture was held for about 3 hours, a second portion of ethyl bromoacetate (0.050 ml, 0.12 equiv.) was added, and the mixture was aged for about 1 hour. The reaction mixture was quenched by excess water (2 ml) to form 5h.
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolanel-1(3bH)-yl)acetate
(5h) from lithium (Z)-2,2,2-trifluoro-1-((1R,5S)-3-oxospiro[bicyclo[3.1.0]hexane-2.2′- [1,3]dithiolanl-4-ylidene)ethan-1-olate (5f)
[00602] A 100 ml flask was charged with ethanol (5 ml). The contents were cooled to about 0 °C and acetyl chloride (1.1 g, 4.0 equiv.) was added over about 10 min. The mixture was agitated at about 0 °C for about 20 minutes and at room temperature for about 20 minutes. To the freshly prepared HCl ethanol solution was added EHA.HCl (0.68 g, 1.2 equiv.) and dithiolane lithium salt 5f (1.0 g). The reaction mixture was heated at about 40 °C for about 22 hours. Ethanol was removed under reduced pressure, and the residue was partitioned between ethyl acetate (5 ml) and water (5 ml). The aqueous layer was discarded, and the organic layer was sequentially washed with aqueous NaHCO3 (5%, 5 ml) and brine (5%, 5 ml) and 5h was
obtained in the EtOAc layer. 1H NMR (400 MHz, DMSO-d6) d 5.14 – 4.97 (m, 2H), 4.14 (qd, J = 7.1, 1.0 Hz, 2H), 3.67 – 3.35 (m, 4H), 2.69 (ddd, J= 8.2, 5.6, 4.2 Hz, 1H), 2.44 (ddd, J= 7.2,
5.5, 3.5 Hz, 1H), 1.37 – 1.29 (m, 1H), 1.21 – 1.14 (m, 3H), 0.44 (ddd, J = 5.3, 4.2, 3.6 Hz, 1H).
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolanel-1 (3bH)-yl)acetate (5h) from (1R,5R)-spiro[bicyclo[3.1.0]hexane-2.2′-[1,3]dithiolanl-3-one (5e)
[00603] 5e (756 mg) was charged to a vessel and dissolved in 2-methyltetrahydrofuran (7.6 mL). To this solution was charged ethyl trifluoroacetate (0.57 g) and the resulting solution was cooled to about 0 °C. Lithium hexamethyldisilazide (1.0 M solution in THF, 4.5 g) was charged over about 60 minutes and reaction was agitated until complete. A solution of sulfuric acid (2.0 g) in water (5.6 mL) was charged, then the reaction was warmed to about 20 °C and agitated for about 20 minutes. Layers were separated and aqueous layer was extracted twice with 2-methyltetrahydrofuran (5.3 mL). Combined organic layer was concentrated to about 0.4 mL and N,N-diisopropylamine (0.5 g) was charged. The product was crystallized by the addition of heptane (11 ml). The slurry was filtered and the filter cake was washed with heptane, then deliquored thoroughly, and dried to afford 5f-01. 1H NMR (400 MHz, Acetonitrile-d3) δ 7.84 (m, 2H), 3.58 (d, J = 8.7 Hz, 2H), 3.47 – 3.27 (m, 4H), 2.20 (s, 1H), 1.81 – 1.68 (m, 1H), 1.24 (dd, J = 6.5, 0.6 Hz, 12H), 0.99 (q, J = 6.5 Hz, 1H), 0.13 (s, 1H).
[00604] Acetyl chloride (1.02 g) was charged to a cooled reaction vessel containing ethanol (5.0 mL) at about 0 °C, then warmed to about 20 °C and agitated for about 30 minutes. In a separate vessel, 5f-01 (1.00 g), ethyl hydrazinoacetate hydrochloride (0.48 g), and lithium chloride (0.39 g) were combined, and the acetyl chloride/ethanol solution was charged to this mixture, followed by tri ethyl orthoformate (1.16 g). The mixture was heated to about 45 °C and agitated until reaction was complete. The reaction was then concentrated to 2 volumes and dichlorom ethane (5.0 mL) was added followed by water (5.0 mL). Layers were separated and organic layer was washed with 5 wt % aqueous sodium bicarbonate followed by 10 wt % aqueous sodium chloride to afford a solution of 5h in dichloromethane that was carried forward into the subsequent step. 1H NMR (400 MHz, DMSO-d6) δ 5.27 – 4.79 (m, 2H), 4.14 (qd, J =
7.1, 1.1 Hz, 2H), 3.70 – 3.42 (m, 4H), 2.68 (dtd, J = 8.0, 6.4, 5.9, 4.4 Hz, 1H), 2.44 (ddd, J = 7.2, 5.5, 3.6 Hz, 1H), 1.32 (ddd, J = 8.2, 7.2, 5.4 Hz, 1H), 1.18 (t, J = 7.1 Hz, 3H), 0.44 (dt, J = 5.4, 3.9 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 167.14, 148.36, 133.80 (q, J = 38.3 Hz), 128.77 (m), 121.54 (q, J = 268.4 Hz), 65.33, 61.79, 51.14, 41.30, 40.98, 40.49, 23.57, 15.52, 14.33; 19F NMR (376 MHz, DMSO-d6) δ -60.31.
Synthesis of (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan]-3-one (5e) from (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan]-3-one (5e) from (1R,5S)-bicyclo[3.1.0]hexan-2-one (4a)
[00605] Tert-butyl nitrite (1.31 g) was charged to a vessel containing 4a (1.00 g, 1.0 equiv) and tetrahydrofuran (5.0 mL) at about 20 °C. Potassium tert-butoxide (6.1 g, 1.7M in tetrahydrofuran) was charged over not less than 30 minutes. The mixture was then agitated until the reaction was complete. The reaction was quenched with aqueous citric acid (2.00 g in 10.00 g water) and extracted with dichloromethane (10.0 mL, 3x). This solution was partially concentrated and the product was isolated by the addition of heptane (6.0 mL). The slurry was filtered and the filter cake was washed with heptane (2.0 mL), then deliquored thoroughly to afford 4d 1H NMR (400 MHz, DMSO-d6) δ 12.26 (s, 1H), 2.73 (d, J = 18.5 Hz, 1H), 2.63 (ddd, J = 18.6, 5.3, 2.0 Hz, 1H), 2.17 – 2.01 (m, 2H), 1.34 (dddd, J= 9.2, 7.1, 4.9, 2.0 Hz, 1H), 0.77 (td, J= 4.6, 3.4 Hz, 1H).
[00606] 1,2-Ethanedithiol (0.41 g) was charged to a vessel containing a solution of 4d (0.50 g, 4.0 mmol) in glacial acetic acid (2.5 mL) at about 20 °C. para-toluenesulfonic acid monohydrate (0.15 g) was added and the mixture was agitated until the reaction was complete. The product was isolated by the addition of water (2 mL). The slurry was filtered and the filter cake was washed with water, then deliquored thoroughly to afford 5i. 1H NMR (400 MHz,
DMSO-d6) δ 10.93 (s, 1H), 3.63 – 3.51 (m, 2H), 3.51 – 3.42 (m, 1H), 3.39 – 3.31 (m, 1H), 2.83 (d, J= 17.4 Hz, 1 H), 2.59 – 2.52 (m, 1H), 1.87 (ddd, J = 8.0, 6.2, 3.7 Hz, 1H), 1.65 (dddd, J=
7.7, 6.2, 5.2, 3.9 Hz, 1H), 0.93 (tdd, J = 7.6, 5.5, 1.7 Hz, 1H), 0.02 (dt, J= 5.5, 3.8 Hz, 1H).
[00607] Para-toluenesulfonic acid (0.90 g) was charged to a vessel containing a suspension of 5i (0.50 g, 2.5 mmol) in methyl ethyl ketone (2.5 mL) and water (2.5 mL). The mixture was agitated at about 85 °C until the reaction was complete. The product was isolated from the reaction mixture by cooling to about 20 °C, adding water (2.50 mL), and cooling to about 0 °C. The slurry was filtered and the filter cake was washed with water, then deliquored thoroughly to afford 5e. 1H NMR (400 MHz, DMSO-d6) δ 3.55 – 3.37 (m, 3H), 3.28 – 3.13 (m, 1H), 3.03 (ddd, J = 18.5, 5.6, 2.2 Hz, 1H), 2.20 (d, J = 18.5 Hz, 1H), 1.84 (ddd, J = 8.0, 7.0, 3.8 Hz, 1H), 1.66 (tdd, J = 7.2, 5.6, 4.1 Hz, 1H), 1.03 (tdd, J = 7.9, 5.9, 2.1 Hz, 1H), 0.06 (dt, J = 6.0, 4.0 Hz, 1H).
Example 6: Preparation of 2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetic acid (VII) from ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane]-1(3bH)-yl)acetate (5h) from ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
[00608] Dichloromethane (27 g) was added to a reactor containing XIV (1.0 g) and cooled to about 10 °C. To this was added 1,2-ethanedithiol (0.18 g, 1.2 equiv.). To this was added boron trifluoride acetic acid complex (3.3 g, 2.5 equivalents) over about 25 minutes, and the reaction mixture was agitated at about 20 °C until complete. A solution of calcium chloride dihydrate (0.80g, 0.78 equiv) in 0.10 N hydrochloric acid (16 g) was added over about 1 hour at about 10 °C, and the mixture was agitated for about 90 minutes at about 20 °C. The organic layer was washed successively with water (8 g) and sodium bicarbonate solution (5 wt/wt%). The organic layer was concentrated to afford 5h. 1H NMR (400 MHz, DMSO-d6) δ 5.27 – 4.79 (m, 2H),
4.14 (qd, J = 7.1, 1.1 Hz, 2H), 3.70 – 3.42 (m, 4H), 2.68 (dtd, J = 8.0, 6.4, 5.9, 4.4 Hz, 1H), 2.44 (ddd, J = 7.2, 5.5, 3.6 Hz, 1H), 1.32 (ddd, J = 8.2, 7.2, 5.4 Hz, 1H), 1.18 (t, J= 7.1 Hz, 3H), 0.44 (dt, J = 5.4, 3.9 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 167. 14, 148.36, 133.80 (q, J= 38.3 Hz), 128.77 (m), 121.54 (q, J= 268.4 Hz), 65.33, 61.79, 51.14, 41.30, 40.98, 40.49, 23.57,
15.52, 14.33. 19F NMR (376 MHz, DMSO-d6) δ -60.31.
Synthesis of ethyl 2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (VII-A)
[00609] Dichloromethane (26 g) was added to a reactor containing 1,3-dibromo-5,5-dimethylhydantoin (DBDMH, 2.4 g, 3.1 equiv.) and cooled to about -10 °C. To this was added 70% hydrofluoric acid/pyridine complex (1.3 g, 17 equiv.), followed by a solution of 5h (1.0 g) in dichloromethane (3 g). The reaction was agitated at about 0 °C until complete. A solution of potassium hydroxide (3.7 g, 25 equivalents) and potassium sulfite (1 .9 g, 4 equiv.) in water (24 g) was added, maintaining an internal temperature of about 5 °C, and agitated for about 30 minutes at about 20 °C. Layers were separated and organic layer was washed with hydrochloric acid (1.1 g, 4 equiv.) in water (9.6 g). The organic layer was concentrated to afford VII-A. 1H NMR (400 MHz, DMSC-d6) δ 5.31 – 5.04 (m, 2H), 4.17 (q, J = 7.1 Hz, 2H), 2.78 – 2.57 (m,
2H), 1.47 (dddd, J = 8.5, 7.1, 5.5, 1.4 Hz, 1H), 1.19 (t, J = 7.1 Hz, 3H), 1.04 (tdt, J= 5.3, 4.0,
1.8 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 166.79, 143.15 (t, J= 29.4 Hz), 134.65 (q, J=
39.0 Hz), 132.99, 121.05 (q, J= 268.4 Hz), 120.52 (t, J= 243.3 Hz), 62.09, 52.49, 27.95 (dd, J = 34.7, 29.0 Hz), 23.82 (d, J = 2.6 Hz), 14.25, 12.14 (t, J = 3.1 Hz). 19F NMR (376 MHz, DMSO-d6) δ -60.47, -79.68 (dd, J= 253.5, 13.2 Hz), -103.09 (dd, J = 253.3, 9.8 Hz).
Synthesis of 2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetic acid (VII)
[00610] A reactor was charged with a solution of VII-A (1.0 g) in dichloromethane (18 g) and cooled to about 5 °C. To this was added ethanol (1.5 g), followed by potassium hydroxide (45 wt/wt%, 0.74 g, 2.0 equiv.). The reaction mixture was agitated at about 20 °C until complete. Water (3.7 g) was added and the reaction mixture was agitated for about 30 minutes. Organic layer was removed and reaction was cooled to about 10 °C. Dichloromethane (18 g) was added, followed by 2N hydrochloric acid (3.3 g, 2,2 equiv.). Reaction was warmed to about 20 °C and agitated for 10 minutes. Layers were separated and aqueous phase was washed with dichloromethane (18 g). Organic layers were combined and concentrated on rotary evaporator to afford VII. 1H NMR (400 MHz, DMSO-d6) δ 13.50 (s, 1H), 5.14 – 4.81 (m, 2H), 2.82 – 2.56 (m, 2H), 1.46 (dddd, J = 8.5, 7.1, 5.5, 1.4 Hz, 1H), 1.08 – 1.00 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 168.16, 143.05 (t, J = 29.4 Hz), 134.40 (q, J = 38.9 Hz), 132.80, 121.11 (q, J = 268.4 Hz), 120.55 (t, J = 243.3 Hz), 52.54, 27.97 (dd, J = 34.7, 29.0 Hz), 23.81 (d, J = 2.5 Hz), 12.13 (t, J = 3.1 Hz). 19F NMR (376 MHz, DMSO-d6) δ -60.39 (d, J = 1.4 Hz), -79.83 (dd, J = 253.2, 13.1 Hz), -102.97 (dd, J= 253.2, 9.8 Hz).
Example 7: Preparation of 4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1- (2,2,2-trifluoroethyl)-1H-indazol-3-amine (V-02) and its mesylated derivatives
Synthesis of 4-chloro-7-bromo-1-(2,2,2-trifluoroethyl)-1H-indazol-3-amine (V-A)
[00611] To a reactor was added tetrahydrofuran (THF, 275 kg) and diisopropyl amine (DIPA, 30 kg) and the mixture was cooled to about -35 °C. nButyl lithium (2.5 mol/L in hexanes, 74 kg) was charged slowly keeping the reaction temperature less than -30 °C. The mixture was agitated at-35 °C until the reaction was complete. 1-bromo-4-chloro-2-fluorobenzene (52 kg) was charged keeping reaction temperature less than 30 °C and the mixture was agitated at -35°C until reaction was complete. N,N-dimethylformamide (DMF, 36 kg) was charged keeping reaction temperature less than -30 °C and the mixture was agitated at about -35 °C until reaction was complete. Hydrochloric acid (HCl, 18 mass% in water, 147 kg) was charged keeping reaction temperature less than -5 °C. The reaction was warmed to about 0 °C, water (312 kg) was added, and the reaction was extracted with methyl tert-butyl ether (MTBE, 770 kg). The organic was warmed to about 20 °C and washed with brine (NaCl, 23.5 mass% in water, 1404 kg). The mixture was distilled to about 3-4 volumes and heptane was charged (354 kg). The product was isolated by distillation to 3-4 volumes. The slurry was filtered and washed with heptane (141 kg) and dried to afford 6a. 1H NMR (400 MHz, DMSO-d6) δ 10.23 (d, J = 1.2 Hz, 1H), 8.00 (dd, J = 8.7, 1.4 Hz, 1H), 7.44 (dd, J = 8.7, 1.4 Hz, 1H).
[00612] 6a (98.5 kg) was charged to a reactor containing acetic anhydride (105 kg) and acetic acid (621 kg) at 20 °C. The mixture was heated to about 45 °C and hydroxyl amine hydrochloride (31.5 kg) was charged. The reaction was heated to about 75 °C and agitated until the reaction was complete. The product was isolated from the reaction mixture by adding water (788 kg) at about 45 °C. The mixture was cooled to about 25 °C and then the slurry was filtered. The filtered cake was washed with water (197 kg,). The cake was dried to afford 6b. 1H NMR (400 MHz, DMSO-d6) δ 8.11 (dd, J= 8.8, 1.4 Hz, 1H), 7.58 (dd, J = 8.8, 1.4 Hz, 1H).
[00613] To a reactor was charged 6b (84 kg), isopropanol (318 kg), and water (285 kg).
Hydrazine hydrate (20 wt% in water, 178 kg) was charged and the mixture was heated to about 80 °C until the reaction was complete. The product was isolated by cooling the reaction to about 25 °C. The slurry was filtered and the filtered cake was washed with a mixture of isopropanol (127 kg) and water (168 kg). The wet solids were recharged to the reactor and water (838 g) was added. The mixture was agitated at about 25 °C and then filtered and washed with water
(168 g, 2 rel). The cake was dried to afford 6c 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 7.41 (d, J= 7.9 Hz, 1H), 6.84 (d, J= 7.9 Hz, 1H), 5.31 (s, 2H).
[00614] 6c (75 kg) was charged to a reactor containing N,N-dimethylformamide (75 kg). Potassium phosphate (99.8 kg) was charged to the reactor at about 25 °C and the mixture was agitated. 2,2,2-trifluoroethyl trifluoromethanesulfonate (74.3 kg) was charged at about 25 °C and the mixture was agitated until the reaction was complete. Water (375 kg) was charged and the mixture was agitated at about 20 °C. The slurry was filtered and washed with water (150 kg). N,N-dimethylformamide (424 kg) and the wet solid were charged to a reactor at about 20 °C.
The mixture was agitated at about 45 °C. 5 % hydrochloric acid (450 kg) was charged drop-wise to the mixture at about 45 °C. The mixture was cooled to about 25 °C. The slurry was filtered and washed with water (375 g). Water (375 kg) and the filtered solid were charged to a reactor at about 20 °C. The mixture was agitated for about 1 hour at about 20 °C. The slurry was filtered and washed with water (375 kg). The cake was dried to afford V-A. 1H NMR (400 MHz, DMSO-d6) δ 7.57 (d, J= 8.1 Hz, 1H), 6.98 (d, J = 8.1 Hz, 1H), 5.70 (s, 2H), 5.32 (q, J = 8.6 Hz,
2H).
Synthesis of 4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2-trifluoroethyl)- 1 H-indazol-3-amine (V-02)
[00615] A reactor containing tetrahydrofuran (27 g) and V-A (1.0 g) was cooled to about 0 °C. Chlorotrimethylsilane (7.6 g, 2.3 equiv) was added, followed by the slow addition of lithium bis(trimethylsilyl)amide (5.7 g, 1 M in THF, 2.1 equiv.). The mixture was stirred at about 0 °C until bistrimethylsilane protection was complete. The solution was washed with ammonium chloride in water (10 wt%, 52 g), toluene (44 g) was added, and the biphasic mixture was filtered through celite. The organic and aqueous phases were separated and the aqueous phase was washed with toluene (44 g). The organics were combined, washed with brine (58 g), and azeotropically distilled . The solution was cooled to about 0 °C, isopropylmagnesium chloride lithium chloride complex (2.7 g, 1.3 M in THF, 1.2 equiv.) was added and the reaction was stirred at about 0 °C until lithium halogen exchange was complete. Isopropoxyboronic acid pinacol ester (6.8 g, 1.2 equiv.) was added and the reaction was stirred at about 0°C until botylation was complete. At about 0 °C, The reaction was quenched with aqueous hydrochloric acid (52 g, 1 M), acetonitrile (16 g) was added, and the mixture was stirred until trimethylsilane deprotection was complete. The solution was extracted with ethyl acetate (45 g) and the organic was washed twice with brine (2 x 58 g). The solution was concentrated to low volumes (26 g), dim ethylformami de (47 g) was added, and the solution was concentrated again (51 g). The product was crystallized by the addition of water (50 g). The slurry was filtered and filter cake was washed with heptane (14 g). The solids were dried to afford V-02. 1H NMR (400 MHz, DMSO-d6) δ 7.70 (dd, J = 7.6, 1.0 Hz, 1H), 7.07 (dd, J = 7.6, 1.0 Hz, 1H), 5.58 (s, 2H), 5.46 (q, J = 9.1Hz, 2H), 1.32 (s, 12H).
Synthesis of 4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2-trifiuoroethyl)- 1 H-indazol-3-amine (V-02)
[00616] To a reactor was charged V-A (30 kg), bis(pinacolato)diboron (27.9 kg), bis(triphenylphosphine)palladium (II) dichloride (0.9 kg, 1.5 mol%), N,N-dimethylformamide (56 kg, 2 rel. vol.) and toluene (157 kg, 6 rel vol.). The mixture was heated to about 105 °C until the reaction was complete. The mixture was cooled to about 25 °C, filtered through celite (15 kg, 0.5 rel. wt.) and rinsed forward with ethyl acetate (270 kg, 10 rel vol.). PSA-17 palladium scavenger (3 kg, 10 wt%) was added and the mixture was stirred at about 45 °C. The mixture was filtered and the cake was washed with ethyl acetate (54 kg, 2 rel. vol.). The mixture was washed twice with lithium chloride (180 kg, 6 rel. vol.) and once with brine (NaCl, 23.5 mass% in water, 180 kg, 6 rel. vol.). The mixture was then concentrated to about 5-6 rel. vol. under vacuum, heated to about 45 °C then cooled to about 25 °C. Heptane (102 kg, 5 rel. vol.) was charged and the mixture was concentrated to about 4-5 rel. vol. The product was isolated by charging heptane (41 kg, 2 rel. vol.) and cooling the mixture to about 0 °C. The slurry was filtered and washed with heptane (41 kg, 2 rel. vol.). The wet solids were recharged to the reactor with ethyl acetate (27 kg, 1 rel. vol.) and heptane (82 kg, 4 rel. vol.), heated to about 65 °C, and then cooled to about 5 °C. The slurry was filtered and washed with heptane (41 kg, 2 rel. vol.). The cake was dried to afford V-02. 1H NMR (400 MHz, DMSO-d6) δ 7.70 (dd, J =
7.6, 1.0 Hz, 1H), 7.07 (dd, J = 7.6, 1.0 Hz, 1H), 5.58 (s, 2H), 5.46 (q, J = 9.1Hz, 2H), 1.32 (s, 12h).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)-N-(methylsulfonyl)methanesulfonamide (V-04)
[00617] To a 100 mL reactor was added V-02 (5.00 g), 2-methyltetrahydrofuran (50 mL), and triethylamine (11.1 mL). The mixture was cooled to about 10 °C and methanesulfonyl chloride (2.58 mL, 33.3 mmol) was added to the mixture. The mixture was agitated at about 10 °C until reaction was complete. The mixture was concentrated to dryness and the residue was purified by column chromatography to afford V-04. 1H NMR (400 MHz, DMSO-d6) δ 7.96 (d, J = 7.7 Hz, 1H), 7.50 (d, J = 7.6 Hz, 1H), 5.95 (q, J = 8.8 Hz, 2H), 3.66 (s, 6H), 1.37 (s, 12H).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2)-1-(2,2,2,- trifluoroethyl)-1H-indazol-3-yl)methanesulfonamide (V-03)
[00618] To a 100 mL reactor was added V-02 (5.00 g), 2-methyltetrahydrofuran (50 mL), and triethylamine (11.1 mL, 79.6 mmol). The mixture was cooled to about 10 °C and methanesulfonyl chloride (2.58 mL) was added to the mixture. The mixture was agitated at about 10 °C until reaction was complete. To the mixture was added 2-methyltetrahydrofuran (21.5 g) and sodium hydroxide (0.43 g) and the mixture was agitated at about 25 °C until the reaction was complete. To the resulting solution was added 2-methyltetrahydrofuran (21.5 g), water (25 g) and acetic acid to achieve a pH of less than 7. The lower aqueous layer was then removed and the organic layer was washed with brine (5 wt%, 7.8g). The organic layer was then concentrated to dryness and the residue was purified by column chromatography to afford V-03. 1H NMR (400 MHz, DMSO-d6) δ 9.96 (s, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.34 (d, J = 7.6 Hz, 1H), 5.80 (q, J = 8.9 Hz, 2H), 3.22 (s, 3H), 1.36 (s, 12H).
Synthesis of N-(7-bromo-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl)-N- (methylsulfonyl)methanesulfonamide (V-06)
[00619] To a reactor was added V-A (3 g), 2-methyltetrahydrofuran (25.8 g), and triethylamine (7.6 mL). The mixture was cooled to about 10 °C, methanesulfonyl chloride (1.8 mL) was added, and the mixture was stirred until reaction was complete. The reaction mixture was washed with aqueous sodium chloride (30 mL) and the organic layer was evaporated to dryness. The residue was purified by column chromatography to afford V-06. 1H NMR (400 MHz, DMSO-d6) δ 7.83 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 5.79 (q, J = 8.5 Hz, 2H), 3.62 (s, 6H).
Synthesis of N-(7-bromo-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl)methanesulfonamide (V-05)
[00620] To a reactor was added V-02 (3 g), 2-methyltetrahydrofuran (30 mL), and triethylamine (7.6 mL). The mixture was cooled to about 10 °C, methanesulfonyl chloride (1.8 mL) was added, and the mixture was stirred until reaction was complete. The reaction mixture was washed with aqueous sodium chloride (30 mL) and the organic portion was concentrated to dryness.
[00621] To the resulting mixture (2.7g) was added 2-methyltetrahydrofuran (15 mL) and sodium hydroxide (1M in water, 15 mL). The mixture was stirred at about 20 °C until the reaction was complete. The aqueous layer was removed and the organic was washed with acetic acid (0.7M in water, 10 mL) and sodium chloride (5 wt% in water, 10 mL).The organic layer was then concentrated to dryness and the residue was purified by column chromatography to afford V-05. 1H NMR (400 MHz, DMSO-D6) δ 10.03 (s, 1H), 7.71 (dd, J = 8.0, 1.6 Hz, 1H), 7.20 (dd, J = 8.1, 1.6 Hz, 1H), 5.64 (q, J = 8.7 Hz, 3H), 3.19 (2, 3H).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2,-dioxaborolan-2-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)-N-(methylsulfonyl)methanesulfonamide (V-04)
[00622] To a reactor was charged V-06 (148 mg), bis(pinacolato)diboron (93 mg), potassium acetate (90 mg) and bis(triphenylphosphine)palladium (II) chloride (4.3 mg, 1.5 mol%). N,N- dimethylformamide (0.2 mL) and toluene (0.6 mL) were added and the reaction was heated to about 105 °C until completion. V-04 was formed. 1H NMR (400 MHz, DMSO-D6) δ 7.96 (d, J = 7.7 Hz, 1H), 7.50 (d, J= 7.6 Hz, 1H), 5.95 (q, J= 8.8 Hz, 2H), 3.66 (s, 6H), 1.37 (s, 12H).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)methanesulfonamide (V-03)
[00623] To a reactor was charged V-05 (124 mg), bis(pinacolato)diboron (93 mg), potassium acetate (90 mg) and bis(triphenylphosphine)palladium (II) chloride (4.3 mg, 1.5 mol%). N,N- dimethylform amide (0.2 mL.) and toluene (0.6 mL, 6 rel. vol.) were added and the reaction was heated to about 105 °C until completion. V-03 was formed. 1H NMR (400 MHz, DMSO-d6) δ
9.96 (s, 1 H), 7.86 (d, J= 7.6 Hz, 1H), 7.34 (d, J= 7.6 Hz, 1H), 5.80 (q, J = 8.9 Hz, 2H), 3.22 (s,
II. Synthesis of the Compound of Formula I
Example 8: Preparation of N-((S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1- yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)- 3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV)
Synthesis of N-((S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2- (3,5-difluorophenyl)ethyl)-2-((3bS.4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro- 1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV) from (S)-1-(3-bromo-6-(3- methyl-3-(methylsulfbnyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3.5-difluorophenyl)ethan-1-amine (VI) Method 1
[00624] n-Propyl phosphonic anhydride (T3P, 3.1 g, 1.5 equiv.) was slowly added to a reactor containing amine VI (1.5 g), acid VII (1.0 g, 1.1 equiv.), triethylamine (Et3N, 0.5 g, 1.5 equiv.), and acetonitrile (MeCN, 8.0 g). The mixture was agitated at about 20 °C until the reaction was complete. The product was crystallized from the reaction mixture with DMF (0.63 g), and water (15 g). The slurry was filtered and the filter cake was washed with a mixture of acetonitrile and water (2 x 2.5 g). The cake was dried to afford IV. 1H NMR (400 MHz, DMSO-d6) δ9.19 (d, J = 8.3 Hz, 1H), 8.12 (d, J = 8.3 Hz, 1H), 7.50 (d, J = 8.3 Hz, 1H), 7.07 (tt, J = 9.4, 2.4 Hz, 1H),
6.96 – 6.87 (m, 2H), 5.52 (td), J = 8.8, 5.3 Hz, 1 H), 4.93 – 4.73 (m, 2H), 3.22 (s, 3H), 3.11 -2.90 (m, 2H), 2.66 – 2.52 (m, 2H), 1.69 (s, 6H), 1.45 – 1.36 (m, 1H), 1.02 – 0.93 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 164.42, 163.62, 163.49, 161.17, 161.04, 158.19, 142.92, 142.20, 142.10, 142.01, 141.63, 140.23, 134.11, 133.73, 132.14, 128.66, 122.23, 120.49, 119.56, 112.49, 112.25, 104.75, 102.25, 88.62, 84.20, 57.44, 53.85, 53.03, 35.21, 23.41, 22.46, 22.40, 11.79.
Synthesis of N-((S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV) from (S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VI) Method 2
[00625] N-methylmorpholine (NMM, 0.51 g, 2.3 equiv.) was added to a vessel containing amine VI (1.0 g), acid VII (1.0 g), 1-hydroxybenzotriazole hydrate (HOBt ● H2O, 0.17 g, 0.5 equiv.), N-(3-dimethylaminopropyi)-N’-ethylcarbodiimide (EDCI ● HCl, 0.52 g, 1.25 equiv.), and acetonitrile (MeCN, 7.8 g). The mixture was agitated at about 20 °C until the reaction was complete. The product was crystallized from the reaction mixture with DMF (2.8 g), and water (10 g). The slurry was filtered and the filter cake was washed with a mixture of acetonitrile and water. The cake was dried to afford IV. 1H NMR (400 MHz, DMSO-d6) δ9.19 (d, J = 8.3 Hz, 1H), 8.12 (d, J = 8.3 Hz, 1H), 7.50 (d, J = 8.3 Hz, 1H), 7.07 (tt, J = 9.4, 2.4 Hz, 1H), 6.96 – 6.87 (m, 2H), 5.52 (td), J = 8.8, 5.3 Hz, 1 H), 4.93 – 4.73 (m, 2H), 3.22 (s, 3H), 3.11 – 2.90 (m, 2H), 2.66 – 2.52 (m, 2H), 1.69 (s, 6H), 1.45 – 1.36 (m, 1H), 1.02 – 0.93 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 164.42, 163.62, 163.49, 161.17, 161.04, 158.19, 142.92, 142.20, 142.10, 142.01, 141.63, 140.23, 134.11, 133.73, 132.14, 128.66, 122.23, 120.49, 119.56, 112.49, 112.25, 104.75, 102.25, 88.62, 84.20, 57.44, 53.85, 53.03, 35.21, 23.41, 22.46, 22.40, 11.79.
Example 9: Preparation of N-((S)-1-(3-(3-amino-4-chloro-1-(2,2,2-trifluoroethyl)-1H- indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5- difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro- 1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (III)
Synthesis of compound III-03
[00626] To a reactor was added IV (1 .0 g), potassium bicarbonate (0.43 g, 1.3 equiv), dichlorobis(tricyclohexylphosphine)palladium(II) (28 mg, 2.5mol%), V-02 (0.67 g), butyl acetate (7.3 g) and water (2.1 g). The reactor was inerted and the mixture was agitated at about 85 °C (75-90 °C) until the reaction was complete. The mixture was cooled to about 40 °C and passed through celite (0.52 g). The celite cake was rinsed with butyl acetate (1.8 g). The filtrate and rinse were combined and this solution was washed twice with a mixture of N-acetyl-L-
cysteine (0.31 g) dissolved in water (5.2 g) and sodium hydroxide in water (5 wt%, 5.4 g). The organics were washed twice with sodium chloride in water (5 wt%, 11 g). The solution was azeotropically distilled into 1-propanol (3.3 g). To the propanol solution at about 50 °C was added methanesulfonic acid (0.31 g, 2.25 equiv.) and the product was crystallized using dibutyl ether (5.1 g). The slurry was cooled to about 10 °C, filtered, and the filter cake was washed with a 5:1 mixture of propanol in dibutyl ether (1.6 g). The solids were dried to afford III-03 1H NMR (400 MHz, DMSO-d6) δ 9.19 (d, J = 8.3 Hz, 2H), 7.84 – 7.69 (m, 4H), 7.11 (d, J = 7.7 Hz, 2H), 7.07 – 6.95 (m, 3H), 6.82 (d, J = 7.7 Hz, 2H), 6.54 – 6.40 (m, 4H), 4.90 (d, J = 16.4 Hz, 2H), 4.76 – 4.60 (m, 4H), 4.15 (dq, J = 16.6, 8.4 Hz, 2H), 3.75 (dt, J = 16.3, 8.7 Hz, 2H), 3.25 (s, 7H), 2.99 – 2.86 (m, 4H), 2.63 – 2.50 (m, 3H), 2.41 (s, 14H), 1.73 (d, J = 2.1 Hz, 13H), 0.93 (dd, J = 6.1, 3.9 Hz, 2H).
Synthesis of N-((S)-1-(3-(3-amino-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (III)
[00627] Aqueous sodium hydroxide (0.2 M; 2.2 equivalents; 9.2 g) was added to a reactor containing III-03 (1.0 g) in MeTHF (8.3 g) at about 20 °C. The biphasic mixture was agitated for about 15 min, and the aqueous layer was removed. The organic layer was washed four times with 2.0 wt% aqueous sodium chloride (9.8 g) and was distilled. The solution containing III was used directly in the II process below. A sample was concentrated to dryness for analysis. 1H NMR (400 MHz, CDCl3): δ 7.44 ( m, 1H), 7.39 (br, 1H), 7.18 (m, 1H), 6.90 (m, 1H), 6.65 (m 1H), 4.10 (m, 2H), 3.72 (m, 4H), 2.78 (m 2H), 2.56 (br, 4H), 1.31 (s, 9H). 13C NMR (100 MHz, DMSO-d6): δ 176.88, 158.95, 141,06, 129.55, 112.79, 109.56, 106.83, 66.66, 65.73, 57.45,
Example 10: Preparation of N-((S)-1-(3-(4-chloro-3-(N- (methylsulfonyl)methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (II)
[00628] Methanesulfonyl chloride (0.32 g, 2.5 equivalents) was added to a reactor containing III (1.0 g), triethylamine (0.69 g, 6.0 equivalents), and MeTHF (11 g) at about 10 °C. The mixture was agitated at about 10 °C until the reaction was complete. The reaction mixture was washed with water (6.4 g) for about 15 minutes, and warmed to about 20 °C. The layers were separated and the organic layer was washed for about 15 minutes with 10 wt% aqueous sodium chloride (6.9 g). The layers were separated and the organic layer was used directly in the next step. An aliquot was concentrated to dryness for analysis. 1H NMR (400 MHz, δ6-DMSO; 9: 1 mixture of atropi somers): δ 9.20 (d, J = 7.9 Hz 1 H), 8.99* (d, J = 8.6 Hz, 1 H), 7.96* (d, J = 7.9 Hz, 1 H), 7.83 (d, J = 8.0 Hz, 1 H), 7.80* (d, J = 7,9 Hz, 1 H), 7.76 (d, J – 8.0 Hz, 1 H), 7.45 (d, J = 7.7 Hz, 1 H), 7.41* (d, J = 7.8 Hz, 1 H), 7.31* (d, J = 7.8 Hz, 1 H), 7.02 (tt, J = 9.4, 2.1 Hz,
1 H), 6.92* (s, 1 H), 6.91 (d, J = 7.7 Hz, 1 H), 6.48 (m, 2 H), 4.92* (s, 1 H), 4.88 (d, J = 16.4 Hz, 1 H), 4.79* (d, J = 16.8 Hz, 1 H), 4.73* (d, J = 16.4 Hz, 1 H), 4.71* (m, 1 H), 4.69 (m, 1 H), 4.62* (s, 1 H), 4.60 (m, 1 H), 4.38* (dq, J = 16.4, 8.2 Hz, 1 H), 4.12 (dq, J = 16.7, 8.4 Hz, 1 H), 3.68* (s, 3 H), 3.66* (s, 3 H), 3.63 (s, 3 H), 3.58 (s, 3 H), 3.26 (s, 3 H), 3.12* (dd, 7 = 13.8, 10.5 Hz, 1 H), 3.05 (dd, J = 13.5, 5.8 Hz, 1 H), 2.97 (dd, J = 13.5, 8.5 Hz, 1 H), 2.78* (dd, J = 13.7, 3.9 Hz, 1 H), 2.59 (m, 1 H), 2.53 (m, 1 H), 1.75 (s), 1.75 (s, 6 H), 1 .39 (m, 1 H), 0.98 (m, 1 H).
13C NMR (100 MHz, DMSO-d6, 9:1 mixture of atropi somers): δ 164.5, 163.6*, 162.1 (dd, ,7 = 246.3, 13.4 Hz), 162.0* (dd, J = 246.1, 13.3 Hz), 158.7, 158.4*, 142.7 (t, J = 29.3 Hz), 142.3, 142.0*, 141.8 (t, J= 9.4 Hz), 140.6*, 139.9, 139.7*, 139.3, 135.8*, 135.0, 133.8 (q, J = 39.0 Hz), 132.2*, 132.1 (m), 131.6, 129.6, 129.4*, 126.7, 125.3, 125.2*, 124.1*, 123.4, 122.8*, 122.7 (q, J= 280.9 Hz), 120.7 (q, J = 268.3 Hz), 119.9 (t, J = 243.7 Hz), 119.8, 119.5*, 119.0*, 118.9, 112.0, 102.2 (t, J= 225.7 Hz), 101.8*, 88.4, 84.5, 57.3, 52.93, 52.86, 52.7, 52.5*, 50.7 (q, J = 33.8 Hz), 50.3*, 42.6*, 42.4, 42.3*, 42.2, 39.51, 39.5, 38.9*, 35.1, 27.5 (dd, J = 35.0, 28.6 Hz), 23.1, 22.4, 22.3, 11.5. (* signals arising from minor atropisomer)
Example 11: Preparation of N-((S)-1-(3-(4-chIoro-3-(methylsuIfonamido)-1-(2,2,2- trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)- 2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5- tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (I)
Synthesis of sodium (4-chloro-7-(2-((S)-1-(2-((3bS.4aR)-5,5-difluoro-3-(trifluoromethyl)- 3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamido)-2-(3,5- difluorophenyl)ethyl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-3-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)(methylsulfonyl)amide (1-02)
[00629] Sodium hydroxide (1 M, 2.9 g, 3.0 equiv.) was added to a reactor containing II (1.0 g) and 2-methyltetrahydrofuran (8.4 g) at about 35 °C. The mixture was agitated until the reaction was deemed complete. The reaction mixture was adjusted to between about 20 and 40 °C and the bottom layer was removed. The organic layer was washed with water (2.9 g) for about 15 minutes, and the bottom layer was removed. The organic solvent was swapped for ethanol and the solution was concentrated to about 5 volumes and the temperature was adjusted to about 35 °C. n-Heptane (3.4 g) was slowly added, and the mixture was aged for about 12 hours. The solids were collected by filtration, and the filter cake was washed with ethanol/n- heptane (1:1). The resultant wet cake was dried under vacuum to afford 1-02. 1H NMR (400 MHz, DMSO-d6) δ 9.09 (d, J = 8.0 Hz, 1H), 8.93* (d, J = 8.5 Hz), 7.80 – 7.72* (m), 7.71 (s, 2H), 6.99 (tt, J = 9.5, 2.4 Hz, 1H), 6.94 (d, J = 7.6 Hz, 1H), 6.90* (d, J = 6.3 Hz), 6.69 (d, J = 7.6 Hz, 1H), 6.57 – 6.51* (m), 6.48 – 6.40 (m, 2H), 4.90 (d, J = 16.5 Hz, 1H), 4.77 (d, J = 16.4
Hz, 1H), 4.70 (td, J = 8.3, 5.2 Hz, 1H), 4.63* (d, J = 16.5 Hz), 4.22 (dq, J= 16.7, 8.4 Hz, 1H), 3.90 – 3.75 (m, 1H), 3.26 (s, 3H), 2.92 (td, J = 13.8, 8.5 Hz, 2H), 2.83* (s), 2.80 (s, 3H), 2.64 – 2.51 (m, 2H), 1.74 (d, J = 2,2 Hz, 6H), 1.44 – 1.34 (m, 1H), 0.94 (dq, J = 6.0, 3.7 Hz, 1H); 13C NMR (100 MHz, dmso) δ 164.39, 163.43, 163.39, 163.25, 160.94, 160.91, 160.81, 158.93,
158.22, 152.64, 151.94, 142.92, 142.72, 142.63, 142.43, 142.34, 142.19, 142.10, 142.00, 141.43,
141.14, 139.55, 139.36, 133.95, 133.56, 133.17, 132.12, 131.93, 131.68, 129.66, 129.56, 128.17,
127.91, 126.86, 126.76, 125.02, 122.35, 122.21, 122.08, 122.05, 119.93, 119.88, 119.38, 118.88,
118.18, 117.54, 117.21, 117.04, 112.18, 112.02, 111.95, 111.84, 111.78, 102.28, 102.03, 101.81,
88.14, 88.00, 84.69, 84.65, 57.33, 53.22, 52.96, 52.76, 52.44, 40.15, 39.94, 39.73, 39.52, 39.31, 39.10, 38.97, 38.89, 38.65, 35.10, 35.08, 27.86, 27.56, 27.52, 27.23, 23.19, 22.42, 22.41, 22.30, 22.28, 11.63. * Signals arising from minor atropisomer. 13C NMR data is reported for the mixture of atropisomers.
Synthesis of N-((S)-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (I) from sodium (4-chioro-7-(2-((S)-1-(2-((3bS.4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-l-yl)acetamido)-2-(3.5-difluorophenyl)ethyl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-3-yl)-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl)(methylsulfonyl)amide (I-02)
[00630] Compound I-02 (1.0 g) and glacial acetic acid (2.1 g) were combined at about 20 °C and were agitated until dissolved. The resultant solution was transferred to a reactor containing water (15 g) over about 1 hour. The resultant slurry was further agitated for about one hour, and was filtered. The wet cake was washed with water (2 x 5 g), deliquored, and dried at about 60 °C under vacuum to provide I. 1H NMR (400 MHz, δ6-DMSO; 5:1 mixture of atropi somers) δ 10.11* (s), 10.00 (s, 1 H), 9.25 (d, J= 8.0 Hz, 1 H), 8.92* (d, J = 8.4 Hz), 7.90* (d, J = 7.6 Hz), 7.81 (d, J = 8.0 Hz, 1 H), 7.76 (d, J= 8.0 Hz, 1 H), 7.32 (d, J = 7.6 Hz, 1 H), 7.23* (d, J = 8.0 Hz), 7.19* (d, J = 8.0 Hz), 7.02 (tt, J = 9.4, 2,4 Hz, 1 H), 6.94* (m), 6.86 (d, J = 7.6 Hz, 1 H), 6.54* (m), 6.48 (m, 2 H), 4.92 (d, J = 16.4 Hz, 1 H), 4.77* (d, J = 16.4 Hz), 4.71 (d, J = 16.4 Hz, 1 H), 4.68* (m), 4.51 (dq, J = 16.4, 8.3 Hz, 1 H), 4.19* (dq, J = 16.4, 8.2 Hz), 3.96 (dq, J = 16.8,
8.4 Hz, 1 H), 3.27 (s, 3 H), 3.24* (s), 3.17 (s, 3 H), 3.11* (dd, J = 13.0, 3.4 Hz), 3.02 (dd, J = 13.6, 5.6 Hz, 1 H), 2.95 (dd, J = 13.8, 8.6 Hz, 1 H), 2.92* (m), 2.60 (m, 1 H), 2.55 (m, 1 H), 1.74 (s, 6 H), 1.40 (m, 1 H), 0.96 (m, 1 H); 13C NMR (100 MHz, δ6-DMSO; 5:1 mixture of atropisomers) δ 164.5, 163.4*, 162.1 (dd, 7 = 246.0, 13.4 Hz), 162.0* (dd, 7 = 246.1, 13.4 Hz), 158.8, 158.1 *, 142.7 (t, 7 = 29.3 Hz), 142.3, 142.1* (m), 141.9 (t, J= 9.5 Hz), 141.7*, 140.2*, 140.0*, 139.8*, 139.5, 139.3, 139.2, 133.8 (q, J= 38.7 Hz), 132.0 (m), 131.7*, 131.1, 130.3*, 130.0, 126.8, 126.4, 126.2*, 123.0* (m), 122.9 (q, J = 281.7 Hz), 122.7*, 122.1, 120.7 (q, J = 268.3 Hz), 119.9 (t, J= 243.4 Hz), 119.0, 118.7*, 117.5*, 117.4, H2.0 (m), 102.1 (t, J= 25.6 Hz), 101.9* (m), 88.5*, 88.4, 84.5, 57.3, 52.8, 52.7, 52.4*, 50.2 (q, J= 33.3 Hz), 50.0 (m),
41.4*, 41.2, 39.8, 38.7, 35.1, 27.5 (dd, J= 35.1, 29.0 Hz), 23.2, 22.4, 22.3, 22.2*, 11.6. * Signals arising from the minor atropisomer.
[00631] Alternatively, a premixed solution of acetic acid (1.5 g), ethanol (12 g), and water (0.3 g) were combined with Compound I-02 at 20 °C and were agitated until dissolved. The resultant solution was transferred to a reactor containing water (100 g) over about 30 minutes. The resultant slurry was further agitated for about one hour, and was filtered. The wet cake was washed with water (2 x 25 g), deliquored, and dried at about 60 °C under vacuum to provide I.
Synthesis of N-((S)-1-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol- 7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,44a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide(I) from N-((S)-1-(3-(3-amino-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)- 3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (III)
[00632] A reactor was charged with III (1.0 g) followed by cyclopentyl methyl ether (2.0 mL). The contents were adjusted to about 80 °C. In a separate reactor, methanesulfonic acid anhydride (0.3g, 1.5 equiv.) was dissolved in cyclopentyl methyl ether (6 mL). The solution was added to the first reactor via a syringe pump over 5 h. Following addition, the reaction mixture was aged for 16 h. The reaction mixture was quenched with water (10 mL). UPLC analysis of the organic phase showed I with 94.8% purity.
Synthesis of N-((S)-1-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol- 7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (I) from N-((S)-1-(3-bromo-6-(3- methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV)
[00633] To a 40 mL vial was added IV (1 .00 g), potassium bicarbonate (420 mg), palladium(II) chloride (4.9 mg, 2.0 mol%), cyclohexyl diphenylphosphine (13.4 mg, 3.6 mol%), V-03 (849 mg), 2-methyltetrahydrofuran (8.0 mL) and water (2.0 mL). The vial was inerted and the mixture was agitated at about 68 °C (65-73 °C) until the reaction was complete. The mixture was cooled to about 40 °C and the aqueous layer was removed. The organic layer was washed with aqueous acetic acid (5% w/v, 5.1 g). The organic was then concentrated to dryness and the residue was purified by column chromatography to afford I. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 0.2H), 10.00 (s, 1H), 9.25 (d, J = 8.2 Hz, 1H), 8.92 (d, J = 8.6 Hz, 0H),
7.90 (d, J = 7.9 Hz, 0.1H), 7.85 – 7.71 (m, 2H), 7.52-7.50 (m, 0.1H), 7.32 (d, J = 7.7 Hz, 1H),
7.21 (q, J= 9.6 Hz, 0.4H), 7.11 – 6.97 (m, 1H), 6.94-6.89 (m, 0.2H), 6.86 (d, J = 7.7 Hz, 1H),
6.55 (d, J = 7.4 Hz, 0.4H), 6.52 – 6.43 (m, 2H), 4.92 (d, J = 16.4 Hz, 1H), 4.81-4.66 (m, 1.5H),
4.64-4.45 (m, 2.4H), 4.28-4.13 (m, 0.2H), 4.08-3.92 (m, 1.6H), 3.32 (s, 0.7H), 3.30-3.22 (m, 4.4H), 3.17 (s, 3H), 3.08-2.89 (m, 2.2H), 2.69 – 2.53 (m, 2.2H), 2.12 (s, 0.2H), 1.99 (s, 1H), 1.91 (s, 0.3H), 1.80 – 1.70 (m, 6H), 1.48-1.36 (m, 1.2H), 1.23 – 1.12 (m, 1.3H), 0.96 (s, 1.2H).
Synthesis of N-((S)-1-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol- 7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3.5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cvclopenta[1,2-c]pyrazol-1-yl)acetamide(I) from N-((S)-1-(3-bromo-6-(3- methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV)
[00634] To a 40 mL vial was added IV (1.00 g), potassium bicarbonate (420 mg), palladium(II) chloride (4.9 mg, 2.0 mol%), cyclohexyl diphenylphosphine (13.4 mg, 3.6 mol%), V-04 (923 mg), 2-methyltetrahydrofuran (8.0 mL) and water (2.0 mL). The vial was inerted and the mixture was agitated at about 68 °C (65-73 °C) until the reaction was complete. The mixture was cooled to about 40 °C and the aqueous layer was removed. The organic was stirred with aqueous sodium hydroxide (5 % w/w, 6.3 g) at 40 °C until reaction was complete. The organic was washed with aqueous acetic acid (5% w/v, 5.1 g). The organic was then concentrated to dryness and the residue was purified by column chromatography to afford I. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 0.2H), 10.00 (s, 1H), 9.25 (d, J = 8.2 Hz, 1H), 8.92 (d, J = 8.6 Hz, 0H), 7.90 (d, J = 7.9 Hz, 0.1H), 7.85 – 7.71 (m, 2H), 7.52-7.50 (m, 0.1H), 7.32 (d, J = 7.7 Hz, 1H), 7.21 (q, J = 9.6 Hz, 0.4H), 7.11 – 6.97 (m, 1H), 6.94-6.89 (m, 0.2H), 6.86 (d, J =
7.7 Hz, 1H), 6.55 (d, J = 7.4 Hz, 0.4H), 6.52 – 6.43 (m, 2H), 4.92 (d, J = 16.4 Hz, 1H), 4.81- 4.66 (m, 1.5H), 4.64-4.45 (m, 2.4H), 4.28-4.13 (m, 0.2H), 4.08-3.92 (m, 1.6H), 3.32 (s, 0.7H), 3.30-3.22 (m, 4.4H), 3.17 (s, 3H), 3.08-2.89 (m, 2.2H), 2.69 – 2.53 (m, 2.2H), 2.12 (s, 0.2H), 1.99 (s, 1H), 1.91 (s, 0.3H), 1.80 – 1.70 (m, 6H), 1.48-1.36 (m, 1.2H), 1.23 – 1.12 (m, 1.3H), 0.96 (s, 1.2H).
SYN
Luíza Cruz
https://drughunter.com/lenacapavir-synthesis-highlights/


see below at end
/////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
 |
|
| Clinical data | |
|---|---|
| Trade names | Sunlenca |
| Other names | GS-CA1, GS-6207 |
| Routes of administration |
By mouth, subcutaneous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| PDB ligand | |
| Chemical and physical data | |
| Formula | C39H32ClF10N7O5S2 |
| Molar mass | 968.28 g·mol−1 |
| 3D model (JSmol) | |
History
Lenacapavir is being developed by Gilead Sciences.[2]
As of 2021, it is in phase II/III clinical trials.[3] It is being investigated as a treatment for HIV patients infected with multidrug-resistant virus and as a twice-yearly injectable for pre-exposure prophylaxis (PrEP).[3][4]
Society and culture
Legal status
On 23 June 2022, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Sunlenca, intended for the treatment of adults with multidrug‑resistant human immunodeficiency virus type 1 (HIV‑1) infection.[5] The applicant for this medicinal product is Gilead Sciences Ireland UC.[5] Lenacapavir was approved for medical use in the European Union in August 2022.[1]
References
- ^ Jump up to:a b c d e f “Sunlenca EPAR”. European Medicines Agency (EMA). 22 June 2022. Archived from the original on 26 August 2022. Retrieved 25 August 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Link JO, Rhee MS, Tse WC, Zheng J, Somoza JR, Rowe W, et al. (August 2020). “Clinical targeting of HIV capsid protein with a long-acting small molecule”. Nature. 584 (7822): 614–618. Bibcode:2020Natur.584..614L. doi:10.1038/s41586-020-2443-1. PMC 8188729. PMID 32612233. S2CID 220293679.
- ^ Jump up to:a b Boerner H (11 March 2021). “Lenacapavir Effective in Multidrug Resistant HIV”. Medscape. Archived from the original on 16 March 2021. Retrieved 15 March 2021.
- ^ Highleyman L (15 March 2021). “Lenacapavir Shows Promise for Long-Acting HIV Treatment and Prevention”. POZ. Archived from the original on 19 July 2021. Retrieved 15 March 2021.
- ^ Jump up to:a b “Sunlenca: Pending EC decision”. European Medicines Agency. 23 June 2022. Archived from the original on 26 June 2022. Retrieved 26 June 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
External links
- “Lenacapavir”. Drug Information Portal. U.S. National Library of Medicine.
- “Lenacapavir sodium”. Drug Information Portal. U.S. National Library of Medicine.
- “Lenacapavir”. Clinical Info. National Institutes of Health.
////////////Lenacapavir sodium, approvals 2022, ema 2022, レナカパビルナトリウム , HIV, SUNLECA, GS-6207, GS-HIV, GS-CA1, GS-CA2, PF-3540074, GS-CA1, eu 2022
[H][C@]12C[C@@]1([H])C(F)(F)C1=C2C(=NN1CC(=O)N[C@@H](CC1=CC(F)=CC(F)=C1)C1=NC(=CC=C1C1=CC=C(Cl)C2=C1N(CC(F)(F)F)N=C2NS(C)(=O)=O)C#CC(C)(C)S(C)(=O)=O)C(F)(F)F
Syn
https://doi.org/10.1021/acs.jmedchem.3c02374
J. Med. Chem. 2024, 67, 4376−4418
Lenacapavir (Sunlenca). Lenacapavir was first approved in the EU in 2022 for use in combination with
other antiretroviral(s) in adults with multidrug resistant HIV infection and for whom it is otherwise not possible to construct a suppressive antiviral regimen.1314Later in the year the drug also received approval in Canada and the US for similar indications.
Lenacapavir received first-in-class designation as 15,16 the first approved drug known to inhibit the capsid of HIV-1, aprotein shell that encompasses the genetic material of the virus and is known to be involved in multiple stages of the HIV life cycle.
Lenacapavir is available as both an oral and injectable therapy. As a slow-release, long lasting treatment
for HIV-1, dosing may be required as infrequently as every 6months when taken in combination with other antiretrovirals, with the goal of improving patient adherence and medication
compliance. Lenacapavir was developed by Gilead Sciences, Inc. and is currently being evaluated in a number of other clinical trials for both HIV-1 treatment and prevention. 13 The synthesis of lenacapavir is achieved by joining four advancedintermediates:17boronateester2.5,18 cyclopropane carboxylic acid 2.13a,17 chiral amino pyridine 2.19,17 and alkynyl sulfone2.20(Schemes2, 3, 4, and5, respectively).18 Thesynthesisof thesekeyintermediateshavebeendescribed inthepatent literatureemployingavarietyof routes17−22and is also exemplified on scales ranging fromsingle gram to multikilogramscale, albeit without reporting isolated yields. For thepurposeof this review, the largest reportedroute to each intermediate will be described. First, synthesis of boronateester2.5beganwitharylcyanofluorideintermediate 2.1 and reliedon a condensation/intramolecular cyclization processwithhydrazinehydrate in i-PrOH/water togenerate
aminopyrazole2.2onmultikilogramscaleafter coolingand precipitation of the product from the reaction mixture (Scheme2).18Next, nucleophilicdisplacementof triflate2.3, facilitated by potassium phosphate in DMF, generated ultimately providing 2.13a as a single isomer to carry forward to lenacapavir. Chiral amine intermediate 2.19 was accessed from 3,6 dibromopicolinaldehyde (2.14), using (S)-2-ethylpropane-2 sulfinamide (2.15) as a chiral auxiliary for installation of the chiral amine (Scheme 4). 17
Condensation of aldehyde 2.14 and sulfinamide 2.15 in the presence of Cs2CO3 and NMP
furnished chiral sulfinimine 2.16, which was precipitated from the reaction mixture by addition of water. The resulting solid was then treated with (3,5-difluorobenzyl)zinc bromide using a controlled addition protocol before HCl-mediated cleavage of the chiral auxiliary. Precipitation from the reaction mixture
yielded the HCl salt of 2.18 as a single isomer, which was treated with Boc2O under aqueous basic conditions to yield the Boc-protected chiral amine 2.19. 17 It is important to note that while this chiral auxiliary-based method constitutes the largest scale route to 2.13a published to date, patent literature
by Gilead has recently emerged, describing synthesis of similar analogs on a small scale using chiral salt-based resolution methods.
The final steps in the synthesis of lenacapavir began with the Sonogashira cross coupling of advanced pyridyl bromide intermediate 2.19 and 3-methyl-3-(methylsulfonyl)but-1-yne (2.20) (Scheme 5). 17
After precipitation from the reaction mixture, the resulting product, 2.21, was subjected to Suzuki
coupling with boronate ester 2.5, furnishing 2.22. Bis sulfonylation of amine 2.22 with MsCl in TEA and subsequent Boc group cleavage with TFA allowed for generation of 2.23 following a hexane wash and neutralization process. Finally, incorporation of the chiral cyclopropane fragment was made
possible by HATU-mediated coupling of amine 2.23 and cyclopropane-carboxylic acid intermediate 2.13a in DMF and DIPEA to give a bis-substituted methylsulfonamide. Selective cleavage of one methanesulfonyl group was performed by addition of 2 N NaOH to the crude reaction mixture,
completing the synthesis of lenacapavir (2).17
(13) Paik, J. Lenacapavir: First approval. Drugs 2022, 82, 1499−
1504.
(14) Tuan, J.; Ogbuagu, O. Lenacapavir: a twice-yearly treatment for
adults with multidrug-resistant HIV infection and limited treatment
options. Expert Rev. Anti Infect. Ther. 2023, 21, 565−570.
(15) Mullard, A. FDA approves first-in-class HIV capsid inhibitor.
Nat. Rev. Drug Discovery 2023, 22, 90.
(16) Dvory-Sobol, H.; Shaik, N.; Callebaut, C.; Rhee, M. S.
Lenacapavir: A first-in-class HIV-1 capsid inhibitor. Curr. Opin. HIV
AIDS 2022, 17, 15−21.
(17) Graupe, M.; Henry, S. J.; Link, J. O.; Rowe, C. W.; Saito, R. D.;
Schroeder, S. D.; Stefanidis, D.; Tse, W. C.; Zhang, J. R. Preparation
of cyclopropacyclopentapyrazolylacetamide compounds useful for the
prophylactic or therapeutic treatment of HIV virus infection. WO
2018035359, 2018.
(18) Allan, K. M.; Batten, A. L.; Brizgys, G.; Dhar, S.; Doxsee, I. J.;
Goldberg, A.; Heumann, L. V.; Huang, Z.; Kadunce, N. T.; Kazerani,
S.; et al. Methods and intermediates for preparation of antiretroviral
pyridine derivative useful for treatment of HIV-1 infections. WO
2019161280, 2019.
.
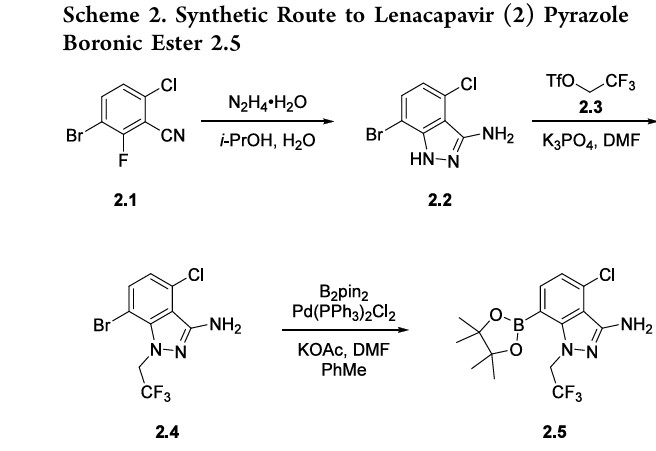



.