















Maribavir
- Molecular FormulaC15H19Cl2N3O4
- Average mass376.235 Da
FDA APROVED 11/23/2021, Livtencity
To treat post-transplant cytomegalovirus (CMV) infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for CMV
Press Release
SYN

1. WO9601833A1.
Syn
US 6204249
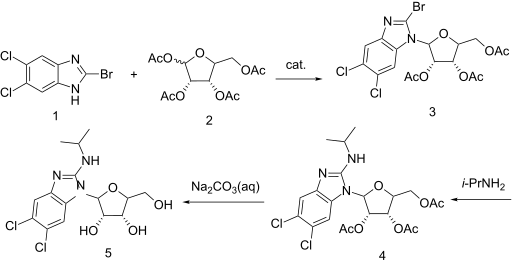
SYN
WO 2001077083
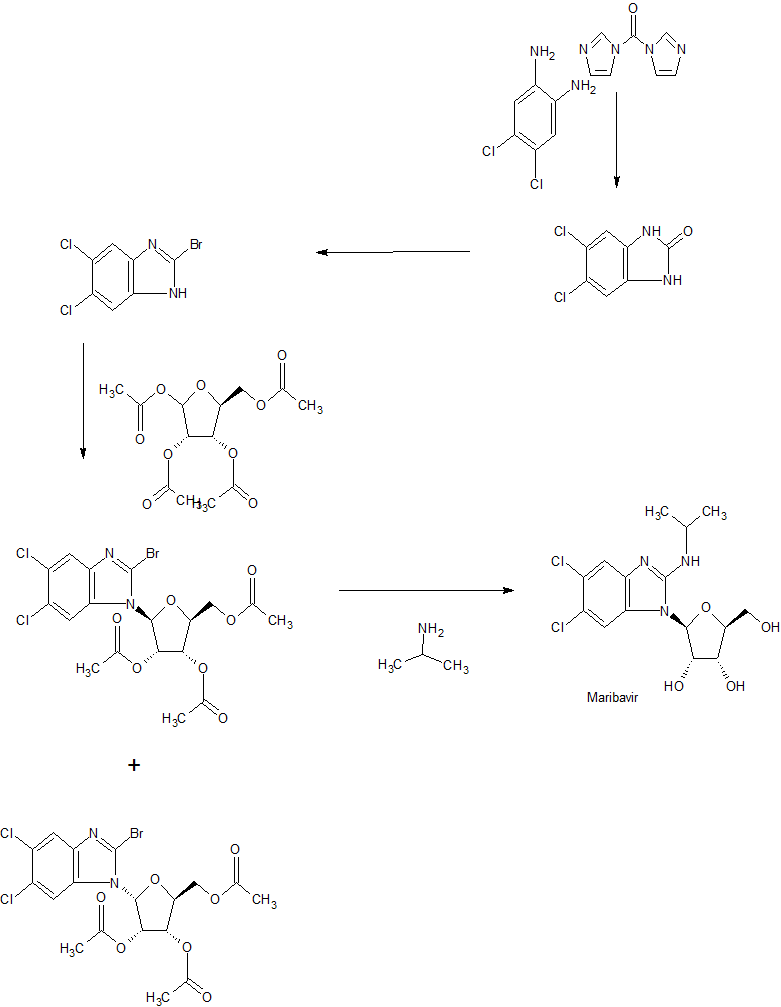
https://patents.google.com/patent/WO2001077083A1/en
H2O: C, 45.70; H, 5.37; N, 10.66. Found: C, 45.75; H, 4.98; N, 10.50.
Maribavir was in phase II clinical trials for the treatment of cytomegalovirus (CMV) infection. It was granted orphan drug designation by the FDA for the indication.
The drug was originally developed by the University of Michigan and was licensed to GlaxoSmithKline. ViroPharma (now subsidiary of Shire) acquired worldwide rights to the drug from GlaxoSmithKline in 2003.
Maribavir, sold under the brand name Livtencity, is an antiviral medication that is used to treat post-transplant cytomegalovirus (CMV).[1][2]
The most common side effects include taste disturbance, nausea, diarrhea, vomiting and fatigue.[2]
Maribavir is a cytomegalovirus pUL97 kinase inhibitor that works by preventing the activity of human cytomegalovirus enzyme pUL97, thus blocking virus replication.[2]
Maribavir was approved for medical use in the United States in November 2021.[2][3]
Medical uses
Maribavir is indicated to treat people twelve years of age and older and weighing at least 35 kilograms (77 lb) with post-transplant cytomegalovirus infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for cytomegalovirus.[2]
Contraindications
Maribavir may reduce the antiviral activity of ganciclovir and valganciclovir, so coadministration with these medications is not recommended.[2]
History
Maribavir is licensed by ViroPharma from GlaxoSmithKline in 2003, for the prevention and treatment of human cytomegalovirus (HCMV) disease in hematopoietic stem cell/bone marrow transplant patients. The mechanism by which maribavir inhibits HCMV replication is by inhibition of an HCMV encoded protein kinase enzyme called UL97 or pUL97.[4] Maribavir showed promise in Phase II clinical trials and was granted fast track status, but failed to meet study goals in a Phase III trial.[5] However, the dosage used in the Phase III trial may have been too low to be efficacious.[6]
A Phase II study with maribavir demonstrated that prophylaxis with maribavir displayed strong antiviral activity, as measured by statistically significant reduction in the rate of reactivation of CMV in recipients of hematopoietic stem cell/bone marrow transplants.[7] In an intent-to-treat analysis of the first 100 days after the transplant, the number of subjects who required pre-emptive anti-CMV therapy was statistically significantly reduced with maribavir compared to placebo.
ViroPharma conducted a Phase III clinical study to evaluate the prophylactic use for the prevention of cytomegalovirus disease in recipients of allogeneic stem cell transplant patients. In February 2009, ViroPharma announced that the Phase III study failed to achieve its goal, showing no significant difference between maribavir and a placebo at reducing the rate at which CMV DNA levels were detected in patients.[8]
The safety and efficacy of maribavir were evaluated in a Phase III, multicenter, open-label, active-controlled trial that compared maribavir with a treatment assigned by a researcher running the study, which could include one or two of the following antivirals used to treat cytomegalovirus: ganciclovir, valganciclovir, foscarnet, or cidofovir.[2] In the study, 352 transplant recipients with cytomegalovirus infections who did not respond (with or without resistance) to treatment randomly received maribavir or treatment assigned by a researcher for up to eight weeks.[2] The study compared the two groups’ plasma cytomegalovirus DNA concentration levels at the end of the study’s eighth week, with efficacy defined as having a level below what is measurable.[2] Of the 235 participants who received maribavir, 56% had levels of cytomegalovirus DNA below what was measurable versus 24% of the 117 participants who received an investigator-assigned treatment.[2]
The U.S. Food and Drug Administration (FDA) granted the application for maribavir orphan drug, breakthrough therapy and priority review designations.[2][3][9][10] The FDA granted the approval of Livtencity to Takeda Pharmaceuticals Company Limited.[2][3]

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
FDA Approves First Treatment for Common Type of Post-Transplant Infection that is Resistant to Other Drugs
Approval is for Cytomegalovirus, a Type of Herpes Virus
- For Immediate Release:
Today, the U.S. Food and Drug Administration approved Livtencity (maribavir) as the first drug for treating adults and pediatric patients (12 years of age and older and weighing at least 35 kilograms) with post-transplant cytomegalovirus (CMV) infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for CMV. Livtencity works by preventing the activity of human cytomegalovirus enzyme pUL97, thus blocking virus replication.
“Transplant recipients are at a much greater risk for complications and death when faced with a cytomegalovirus infection,” said John Farley, M.D., M.P.H., director of the Office of Infectious Diseases in the FDA’s Center for Drug Evaluation and Research. “Cytomegalovirus infections that are resistant or do not respond to available drugs are of even greater concern. Today’s approval helps meet a significant unmet medical need by providing a treatment option for this patient population.”
CMV is a type of herpes virus that commonly causes infection in patients after a stem cell or organ transplant. CMV infection can lead to CMV disease and have a major negative impact on transplant recipients, including loss of the transplanted organ and death.
Livtencity’s safety and efficacy were evaluated in a Phase 3, multicenter, open-label, active-controlled trial that compared Livtencity with a treatment assigned by a researcher running the study, which could include one or two of the following antivirals used to treat CMV: ganciclovir, valganciclovir, foscarnet or cidofovir. In the study, 352 transplant recipients with CMV infections who did not respond (with or without resistance) to treatment randomly received Livtencity or treatment assigned by a researcher for up to eight weeks.
The study compared the two groups’ plasma CMV DNA concentration levels at the end of the study’s eighth week, with efficacy defined as having a level below what is measurable. Of the 235 patients who received Livtencity, 56% had levels of CMV DNA below what was measurable versus 24% of the 117 patients who received an investigator-assigned treatment.
The most common side effects of Livtencity include taste disturbance, nausea, diarrhea, vomiting and fatigue. Livtencity may reduce the antiviral activity of ganciclovir and valganciclovir, so coadministration with these drugs is not recommended. Virologic failure due to resistance can occur during and after treatment with Livtencity, therefore CMV DNA levels should be monitored and Livtencity resistance should be checked if the patient is not responding to treatment or relapses.
Livtencity received Breakthrough Therapy and Priority Review designations for this indication. Breakthrough Therapy designation is a process designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s). Priority Review designation directs overall attention and resources to the evaluation of applications for drugs that, if approved, would be significant improvements in the safety or effectiveness of the treatment, diagnosis or prevention of serious conditions when compared to standard applications.
The FDA granted the approval of Livtencity to Takeda Pharmaceuticals Company Limited.
Related Information
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215596lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l m “FDA Approves First Treatment for Common Type of Post-Transplant Infection that is Resistant to Other Drugs”. U.S. Food and Drug Administration (FDA) (Press release). 23 November 2021. Retrieved 23 November 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c “Takeda’s Livtencity (maribavir) Approved by U.S. FDA as the First and Only Treatment for People Ages 12 and Older with Post-Transplant Cytomegalovirus (CMV), Refractory (With or Without Genotypic Resistance) to Conventional Antiviral Therapies”. Takeda (Press release). 23 November 2021. Retrieved 26 November 2021.
- ^ Biron KK, Harvey RJ, Chamberlain SC, Good SS, Smith AA, Davis MG, et al. (August 2002). “Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a benzimidazole L-riboside with a unique mode of action”. Antimicrobial Agents and Chemotherapy. 46 (8): 2365–72. doi:10.1128/aac.46.8.2365-2372.2002. PMC 127361. PMID 12121906.
- ^ Marty FM, Ljungman P, Papanicolaou GA, Winston DJ, Chemaly RF, Strasfeld L, et al. (April 2011). “Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem-cell transplants: a phase 3, double-blind, placebo-controlled, randomised trial”. The Lancet. Infectious Diseases. 11 (4): 284–92. doi:10.1016/S1473-3099(11)70024-X. PMID 21414843.
- ^ Snydman DR (April 2011). “Why did maribavir fail in stem-cell transplants?”. The Lancet. Infectious Diseases. 11 (4): 255–7. doi:10.1016/S1473-3099(11)70033-0. PMID 21414844.
- ^ Phase 2 Data Shows Maribavir Markedly Reduced Rate Of Cytomegalovirus Infection And Disease In Bone Marrow Transplant Patients, Medical News Today, Jun 2, 2008
- ^ ViroPharma:Maribavir Phase III Study Missed Goal;Shares Plunge, CNN Money, February 09, 2009
- ^ “Maribavir Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 1 February 2007. Retrieved 26 November 2021.
- ^ “Maribavir Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 7 June 2011. Retrieved 26 November 2021.
External links
- “Maribavir”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02931539 for “Efficacy and Safety Study of Maribavir Treatment Compared to Investigator-assigned Treatment in Transplant Recipients With Cytomegalovirus (CMV) Infections That Are Refractory or Resistant to Treatment With Ganciclovir, Valganciclovir, Foscarnet, or Cidofovir” at ClinicalTrials.gov
 |
|
| Clinical data | |
|---|---|
| Trade names | Livtencity |
| Other names | 1263W94 |
| License data | |
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChemCID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| NIAID ChemDB | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C15H19Cl2N3O4 |
| Molar mass | 376.23 g·mol−1 |
| 3D model (JSmol) | |
| |
|
/////////Maribavir, APPROVALS 2021, FDA 2021, Livtencity, Takeda, Breakthrough Therapy, Priority Review , ORPHAN, UNII-PTB4X93HE1, марибавир , ماريبافير ,马立巴韦 , BW-1263W94, Camvia, D04859, G1263, GW257406X, 1263W94, BW-1263W94, GW-1263, GW-257406X, SHP-620, VP-41263,