GUANGZHOU, China–(BUSINESS WIRE)–MaxiNovel Pharmaceuticals, Inc. announced today that the U.S. Food and Drug Administration (“FDA”) has granted MaxiNovel Orphan Drug Designation for MAX-40279 in the treatment of Acute Myeloid Leukemia (AML).
AML is the most common acute leukemia which accounts for approximately 25% of all adult leukemias worldwide. Approximately one-third of AML patients have a FLT3 gene mutation. Such mutation can result in faster disease progression, higher relapse rates and lower rates of survival than other forms of AML. Inhibition of FLT3 mutation is of high importance in combating AML.
In the preclinical testing, MAX-40279 demonstrated potent inhibition of both FLT3 and FGFR with excellent drug concentration in the bone marrow. It is designed to overcome the observed drug resistance of the current FLT3 inhibitors due to the bone marrow FGF/FGFR pathway activation.
“We are very pleased to receive the ODD,” commented MaxiNovel’s Vice President Dr. Elizabeth Ashraf. “Our objective is to bring the best in class medicine to the patients worldwide.”
The FDA Office of Orphan Products Development grants orphan drug designation to novel drugs and biologics that are intended for the safe and effective treatment, diagnosis or prevention of rare diseases or disorders that affect fewer than 200,000 people in the United States. The designation allows manufacturers to qualify for various incentives including federal grants, tax credits for qualified clinical trials, a waiver of PDUFA filing fees and 7 years of market exclusivity upon regulatory approval.
About MaxiNovel Pharmaceuticals, Inc:
Maxinovel Pharmaceuticals, Inc. is a biotech company focusing on the discovery and development of Immuno-oncology therapy and targeted therapy. It will use its orally active Immuno-oncology product platform to bring effective combo product of multi-components in a single oral pill to the patients worldwide. For more info: www.maxinovel.com
The JAK-STAT (Janus kinase-signal transducer and activator of transcription) signal pathway is a signal transduction pathway stimulated by cytokines discovered in recent years, and it participates in many important biology such as cell proliferation, differentiation, apoptosis and immune regulation. Process (Aaronson, D Set al. Science 2002, 296, 1653-1655; O’Shea, J Jet al. Nat. Rev. Drug Discovery 2004, 3, 555-564). Compared with other signal pathways, the transmission process of this signal pathway is relatively simple. It mainly consists of three components, namely tyrosine kinase-related receptor, tyrosine kinase JAK and transcription factor STAT. JAK (Janus Kinase), a type of molecule in the cell, is rapidly recruited and activated on the receptor after receiving the signal from the upstream receptor molecule. The activated JAK catalyzes the receptor tyrosine phosphorylation, and the phosphorylation of tyrosine on the receptor molecule Amino acid is the recognition and binding site of a kind of signal molecule STAT SH2. Tyrosine phosphorylation occurs after STAT binds to the receptor. Tyrosine phosphorylated STAT forms a dimer and enters the nucleus. As an active transcription factor, dimeric STAT molecules directly affect the expression of related genes, thereby changing the proliferation or differentiation status of target cells.
The JAK-STAT pathway is widely present in various tissues and cells in the body, and has an important role in the differentiation, proliferation, and anti-infection of lymphocytes, and participates in the interaction of various inflammatory factors and signal transduction (Kiesseleva T. et al. . J. Gene, 2002, 285, 1-24). The abnormal activation of this pathway is closely related to a variety of diseases. Finding and screening JAK inhibitors can help in-depth study of the regulatory mechanism of JAK-STAT, thereby providing new drugs and methods for the prevention and treatment of related diseases
The occurrence, growth, invasion and metastasis of tumors are related to the JAK-STAT signal transduction pathway. In normal signal transduction, the activation of STATs is rapid and transient. The continuous activation of STATs is closely related to the process of malignant transformation of cells (Buettner R. et al. Clin. Cancer Res. 2002, 8(4), 945-954). STAT3 is the focus of multiple oncogenic tyrosine kinase signal channels such as EGFR, IL-6/JAK, Src, etc. It is activated in a variety of tumor cells and tissues, such as breast cancer, ovarian cancer, and head and neck squamous cells. Like cell carcinoma, prostate cancer, malignant melanoma, multiple myeloma, lymphoma, brain tumor, non-small cell lung cancer and various leukemias, etc. (Niu G. et al. Oncogene 2002, 21(13), 2000-2008 ). JAK-STAT pathway inhibitors belong to PTK inhibitors, and this enzyme is a member of the oncogene protein and proto-oncoprotein family, and plays an important role in the normal and abnormal cell proliferation. The occurrence and growth of tumors are inseparable from PTK. Therefore, JAK-STAT pathway inhibitors inhibit tumor growth by antagonizing PTK, and have obvious anti-tumor effects (Mora LBet al.J.Cancer Res.2002,62(22) , 6659-6666).
In addition, the latest research shows that: organ transplant rejection, psoriasis, tissue and organ fibrosis, bronchial asthma, ischemic cardiomyopathy, heart failure, myocardial infarction, blood system diseases, and immune system diseases are all related to JAK-STAT signaling. The pathway is closely related. This signaling pathway is not only important for maintaining the normal physiological functions of cells, but also has an important regulatory role for the occurrence and development of diseases.
The Fibroblast Growth Factor Receptor family belongs to a new type of receptor kinase family, which includes four receptor subtypes (FGFR-1,2,3) encoded by four closely related genes. And 4) and some heterogeneous molecules, which form a ternary complex with fibroblast growth factor (FGF) and heparan sulfate, and then trigger a series of signal transduction pathways to participate in the regulation of physiological processes in the organism. FGFR has a wide range of physiological and pathological effects in the body: (1) Embryo development. Studies have shown that during embryonic development, FGFR signal transduction is essential for most organ development and the formation of embryonic patterns. (2) Cell division, migration and differentiation. FGFR stimulates cell proliferation and participates in the regulation of cell transformation in the pathological process. There are many parallel pathways to achieve FGFR-mediated cell division signal transduction, which has been confirmed by many studies (JKWang et al., Oncogene 1997, 14, 1767 -1778.). (3) Bone diseases. The growth and differentiation of bones are also regulated by the FGF family, and mutations in FGFR can cause bone deformities (R. Shang et al., Cell 1994, 78, 335-342.). (4) The occurrence of tumors. FGFR can promote the migration, proliferation and differentiation of endothelial cells, and plays an important role in the regulation of angiogenesis and angiogenesis. Uncontrolled angiogenesis can lead to the occurrence of tumors and the growth of metastases (J.Folkman.Nat.Med.1995) ,1,27-31.).
FMS-like tyrosine kinase 3 (FMS-like tyrosine kinase 3, FLT3) belongs to the type III receptor tyrosine kinase (receptor tyrosine kinase III, RTK III) family member, it is composed of extracellular domain, intracellular domain and The transmembrane region is composed of 3 parts, which are first expressed in human hematopoietic stem cells. FLT3 interacts with its ligand FL to stimulate or act on stem cells, which is of great significance to the growth and differentiation of stem cells. FLT3 kinase has wild-type FLT3-WT and its main activation mutant FLT3-ITD and FLT3-D835Y. FLT3 is mainly expressed in the precursors of normal myeloid cells, but its abnormal expression is also found in a large part of acute myeloid leukemia (AML) cells.
In recent years, many large-scale studies have confirmed that activating mutations of FLT3 play a very important pathological role in the occurrence and progression of acute myeloid leukemia. FLT3 has become an important target for the treatment of acute myeloid leukemia.
rc family kinase (SFK) is a family of non-receptor tyrosine kinases, including c-Src, LYN, FYN, LCK, HCK, FGR, BLK, YES and YRK, among which LYN kinase has LYNα and LYNβ Both subtypes, LYN kinase and its two subtypes can cause similar intracellular tyrosine phosphorylation. According to the amino acid sequence, SFK can be divided into two sub-families: one family is c-Src, FYN, YES and FGR, which are widely expressed in different tissues; the other family is LCK, BLK, LYN and HCK, which are closely related to hematopoietic cells. SFK is connected to multiple signal transduction pathways in the body, and can be activated by growth factors, cytokines and immune cell receptors, G protein-coupled receptors, integrins and other cell adhesion molecules, and then activate the corresponding signal transduction pathways , Causing a variety of physiological effects of cells. The activity of SFK mainly includes the regulation of cell morphology, cell movement, cell proliferation and survival. The abnormal activation and expression of these kinases leads to the occurrence and development of a wide range of diseases, such as a large number of solid tumors, various hematological malignancies and some neuronal pathologies. Therefore, looking for SFK inhibitors is a promising research topic in the field of medicinal chemistry.
2,4-Dichloro-6-methylthiophene [3,2-d] pyrimidine (10g, 45.6mmol) was dissolved in tetrahydrofuran (100mL) and ethanol (100mL), and the reaction solution was cooled to 0°C and divided Sodium borohydride (12.5 g, 198 mmol) was added in batches. The reaction solution was raised to room temperature and continued to stir for 16 hours, diluted with water (500 mL), and then adjusted to pH=7 with 1N aqueous hydrochloric acid. The aqueous phase was extracted with ethyl acetate (150 mL×3). The organic phase was washed sequentially with water (100mL×3) and saturated brine (100mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain a white solid 31-e (7.5g, yield: 88%). The product does not require further purification. LC-MS(ESI): m/z=187[M+H] + .
[0492]
Synthesis of compound 31-d
[0493]
Compound 31-e (7.5 g, 40 mmol) was dissolved in chloroform (300 mL) at 0°C, active manganese dioxide (35 g, 400 mmol) was added, the reaction solution was raised to room temperature and stirring was continued for 16 hours. The reaction solution was filtered through Celite, and the filter cake was washed with chloroform (100 mL×3). The combined filtrates were concentrated under reduced pressure to obtain white solid 31-d (6.6 g, yield: 89%), which did not require further purification. LC-MS(ESI): m/z=185[M+H]+.
[0494]
Synthesis of compound 31-c
[0495]
Compound 31-d (3.1g, 16.8mmol) was dissolved in trifluoroacetic acid (30mL) at 0℃, N-iodosuccinimide (5.7g, 25.3mmol) was added in batches, and the reaction solution was raised to Keep stirring at room temperature for 1 hour. Water (50 mL) was added to the reaction solution to quench the reaction, and it was extracted with dichloromethane (50 mL×3). The organic phase was washed successively with water (50mL×3) and saturated brine (50mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain a white solid 31-c (4.9g, yield: 94%). The product does not require further purification. LC-MS(ESI): m/z=311[M+H] + .
[0496]
Synthesis of compound 31-b
[0497]
Compound 31-c (615mg, 1.98mmol), 2-methoxy-4-fluorophenylboronic acid (405mg, 2.38mmol) and sodium carbonate (630mg, 5.94mmol) were suspended in dioxane (5mL) water (5mL) ), add [1,1′-bis(diphenylphosphorus)ferrocene]dichloropalladium dichloromethane complex (163mg, 0.2mmol). Replace with nitrogen 3 times, and heat to 80°C to react for 16 hours. After cooling to room temperature, the reaction solution was concentrated under reduced pressure. The residue was partitioned with dichloromethane (50mL) and water (50mL). The organic phase was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography (petroleum Ether: dichloromethane=1:1) to obtain a white solid 31-b (240 mg, yield: 39%). LC-MS(ESI): m/z=309[M+H] + .
[0498]
Synthesis of compound 31-a
[0499]
Compound 31-b (240mg, 0.78mmol) and compound 32-c (208mg, 0.78mmol) were dissolved in N,N-dimethylformamide (3mL), potassium carbonate (323mg, 2.34mmol) was added, 2- Dicyclohexylphosphine-2′,6′-diisopropoxy-1,1′-biphenyl (112 mg, 0.24 mmol) and tris(dibenzylideneacetone) dipalladium (134 mg, 0.24 mmol). Under the protection of nitrogen, heat to 110°C to react for 16 hours. After cooling to room temperature, the reaction solution was partitioned with dichloromethane (50 mL) and water (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel thin layer chromatography preparation plate (petroleum Ether: ethyl acetate = 1:1) to obtain a yellow viscous oil 31-a (190 mg, yield: 45%). LC-MS(ESI): m/z=539[M+H] + .
[0500]
Synthesis of compound 31
[0501]
31-a (190 mg, 0.35 mmol) was dissolved in dichloromethane (3 mL), trifluoroacetic acid (3 mL) was added, and the mixture was stirred at room temperature for 3 hours. The reaction solution was concentrated under reduced pressure. The residue was layered with ethyl acetate (50mL) and 1N aqueous hydrochloric acid (50mL). The aqueous phase was adjusted to pH=10 with saturated aqueous potassium carbonate solution. 3) Washing and vacuum drying the solid to obtain a light yellow solid 31 (22 mg, yield: 14%). LC-MS(ESI): m/z=439[M+H] + .
Example 1 Preparation of fumarate of fused ring pyrimidine compound as shown in formula 2
Weigh the compound N-[7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-d]pyrimidin-2-yl]-1-(piperidine-4- Base)-1H-pyrazol-4-amine (synthesized according to Example 31 of patent CN106366093A) 100mg (0.228mmol, 1eq) into the vial, add 10mL 88% acetone-water solution, add the vial at about 50°C and stir until dissolved clear. 1.1 mL of fumaric acid with a concentration of 0.25 mol/L in ethanol (0.275 mmol, 1.2 eq) was slowly added dropwise to the free base solution of fused ring pyrimidine compounds, and stirred at 50 ℃ for 1 hour, and then the solution was The rate of 5°C/h was slowly reduced to room temperature, and the solid was collected and dried under vacuum at 40°C overnight.
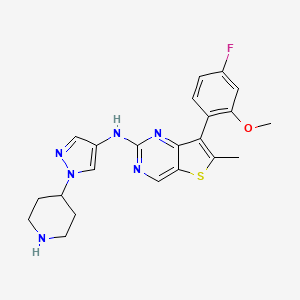
7-(4-Fluoro-2-methoxyphenyl)-6-methyl-N-(1-piperidin-4-yl)-1hydro-pyrazol-4-yl)thieno[3,2 -D]pyrimidine-2-amino is a strong JAK, FGFR, FLT3 kinase inhibitor, and has a good application prospect in the treatment of tumors, immune system diseases, allergic diseases and cardiovascular diseases. This compound is described in patent CN106366093A and has the following chemical structure:
CN106366093A discloses the preparation method of the compound:
In the above synthetic route, NaBH 4 is sodium borohydride, MnO 2 is manganese dioxide, NIS is N-iodosuccinimide, TFA is trifluoroacetic acid, and Pd(dppf)Cl 2 is [1,1′- Bis(diphenylphosphino)ferrocene]palladium dichloride, DIAD is diisopropyl azodicarboxylate, PPh 3 is triphenylphosphine, Pd/C is palladium on carbon, Pd 2 (dba) 3 is Tris(dibenzylideneacetone)dipalladium, RuPhos is 2-bicyclohexylphosphine-2′,6′-diisopropoxybiphenyl.
However, the above method has the problems of a large number of reaction steps, low yield, and requires column chromatography for separation and purification, and is not suitable for industrial scale-up production. Therefore, it is necessary to improve its preparation method.
The present invention provides a method for preparing a compound represented by formula B, which comprises the following steps: under a protective gas atmosphere, in a solvent, in the presence of a catalyst and a base, a compound represented by formula C is combined with a compound represented by formula K The compound can be subjected to the coupling reaction shown below; the catalyst includes a palladium compound and a phosphine ligand;
The preparation method of the compound represented by formula B may further include the following steps: in an organic solvent, in the presence of a base, the compound represented by formula E and the compound represented by formula D are subjected to the substitution reaction shown below, To obtain the compound represented by formula C;
The present invention provides a method for preparing a compound represented by formula C, which comprises the following steps: in an organic solvent, in the presence of a base, a compound represented by formula E and a compound represented by formula D are subjected to the following steps: Substitution reaction is enough;
Example 1: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
Into a 500L reactor, add 10% palladium on carbon (4.6Kg), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (24.2Kg, 109.5mol), and tetrahydrofuran (150Kg) in sequence And N,N-diisopropylethylamine (17.0Kg, 131.5mol). Fill the kettle with hydrogen, and control the hydrogen pressure at 0.5 MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 120 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (58Kg) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (60Kg) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 360Kg of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was separated out, centrifuged, and the filter cake was vacuum dried to obtain the product 2-chloro-6-methylthieno[3,2-D]pyrimidine 18.94Kg, yield: 93.2%. LC-MS(ESI): m/z=185.1[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.30 (s, 1H), 7.34 (s, 1H), 2.73 (s, 3H).
Example 2: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
To a 100mL reaction flask, add 10% palladium on carbon (0.17g), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (2g, 9.2mmol), tetrahydrofuran (40mL) and N,N-Diisopropylethylamine (1.412 g, 10.9 mmol). Fill the bottle with hydrogen and control the hydrogen pressure at 0.5MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 20 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (2.1 g) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (2.2g) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 13.3g of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was precipitated, centrifuged, and the filter cake was vacuum dried to obtain 2.4 g of 2-chloro-6-methylthieno[3,2-D]pyrimidine as a product, with a yield of 82%. The LC-MS and 1 H NMR are the same as in Example 1.
Example 3: 7-Bromo 2-chloro-6-methylthieno[3,2-D]pyrimidine (Compound E)
Add trifluoroacetic acid (150Kg) and 2-chloro-6-methylthieno[3,2-D]pyrimidine (18.90Kg, 102.4mol) into a 500L enamel reactor. Add N-bromosuccinimide (18.33Kg, 103.0mol) under temperature control at 15±5℃. After the addition, the temperature is controlled at 25±5℃ to react for 2 hours. Sampling to monitor the reaction, there is still a small amount of raw materials remaining. Additional N-bromosuccinimide (1.0 Kg, 5.6 mol) was added, stirring was continued for 1 hour, sampling and monitoring showed that the reaction was complete. Control the temperature at 10±5°C, and add 274Kg of water dropwise. After the addition, stir at 10±5°C for 2 hours. After centrifugation, the solid was vacuum-dried to obtain the product, 7-bromo-2-chloro-6-methylthieno[3,2-D]pyrimidine, 24.68Kg, yield: 91.4%. LC-MS(ESI): m/z=265.0[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.33 (s, 1H), 2.64 (s, 3H).
Example 4: 4-(p-toluenesulfonyl)-piperidine-1-tert-butyl carbonate (Compound G)
Add pyridine (176Kg) and N-BOC-4-hydroxypiperidine (36.00Kg, 178.9mol) to a 500L enamel reactor. Add p-toluenesulfonyl chloride (50.5Kg, 264.9mol) in batches under temperature control at 10±10°C. After the addition, the temperature is controlled at 25±5°C to react for 18 hours. The reaction solution was transferred to a 1000L reactor, the temperature was controlled at 15±5°C, and 710Kg of water was added dropwise. After the addition, stir at 15±5°C for 2 hours. After filtration, the solid was washed with water and dried in vacuum to obtain the product 4-(p-methylbenzenesulfonyl)-piperidine-1-carbonate tert-butyl ester, 59.3Kg, yield: 93.3%. LC-MS(ESI): m/z=378.0[M+Na] + .
Example 5: 4-(4-Nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound F)
Add N,N-dimethylformamide (252Kg), 4-(p-methylbenzenesulfonyl)-piperidine-1-carbonate tert-butyl ester (59.3Kg, 166.8mol), 4-nitro to the reaction kettle Pyrazole (21.5Kg, 190.1mol), and anhydrous potassium carbonate (34.3Kg, 248.2mol). The temperature was controlled at 80±5°C and the reaction was stirred for 18 hours. Cool down to 15±5°C, add 900Kg of water dropwise, control the dropping rate, and keep the temperature at 15±5°C. After the addition, stir at 5±5°C for 2 hours. After filtering, the solid was washed twice with water and dried in vacuum to obtain the product 4-(4-nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 39.92Kg, yield: 80.8%. LC-MS (ESI): m/z=319.1 [M+Na] + .
1 H NMR (400MHz, d 6 -DMSO): δ8.96(s,1H), 8.27(s,1H), 4.44-4.51(m,1H), 4.06-4.08(m,2H), 2.75-2.91( m, 2H), 2.04-2.07 (m, 2H), 1.80-1.89 (m, 2H), 1.41 (s, 9H).
Example 6: 4-(4-Amino-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound D)
Add 10% palladium-carbon (2.00Kg), 4-(4-nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (39.94Kg, 134.09mol) to the reaction kettle, nothing Water ethanol (314Kg) and ammonia (20.0Kg, 134.09mol). Fill the kettle with hydrogen, and control the hydrogen pressure at 0.2MPa. Turn on the stirring and keep the temperature at 45±5°C to react for 4 hours. Filter, collect the filtrate, and concentrate the filtrate under reduced pressure. Add ethyl acetate (40Kg) and n-heptane (142Kg) to the concentrate, stir at 25±5°C for 1 hour, and then lower the temperature to 5±5°C and stir for 2 hours. After filtration, the solid was vacuum dried to obtain the product 4-(4-amino-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 31.85Kg, yield: 88.6%. LC-MS(ESI): m/z=267.2[M+H] + .
Example 7: 4-(4-(7-Bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1hydro-pyrazol-1-yl)piperidine-1 -Tert-butyl carbonate (compound C)
Add n-butanol (117Kg), N,N-diisopropylethylamine (15.00Kg, 116.06mol), 4-(4-amino-1hydro-pyrazol-1-yl)piperidine to the reaction kettle 1-tert-butyl carbonate (32.02Kg, 120.22mol) and 7-bromo-2-chloro-6-methylthieno[3,2-D]pyrimidine (24.68Kg, 93.65mol). Turn on the stirring and keep the temperature at 100±5°C to react for 42 hours. Concentrate under reduced pressure. Methanol was added to the concentrate to be beaten. The solid was filtered and dried under vacuum to obtain the product 4-(4-(7-bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1hydro-pyrazol-1-yl ) Piperidine-1-tert-butyl carbonate 37.26Kg, yield: 80.6%. LC-MS(ESI): m/z=493.1[M+H] + .
Example 8: 4-(4-((7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1 Hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound B)
Add purified water (113Kg), dioxane (390Kg), 4-(4-(7-bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino) into the reactor -1H-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (37.26Kg, 93.65mol), 2-methoxy-4-fluorophenylboronic acid pinacol ester (23.05Kg, 120.22mol) , Anhydrous potassium carbonate (20.95Kg, 151.8mol), palladium acetate (0.18Kg, 0.80mol) and 2-dicyclohexylphosphine-2,4,6-triisopropylbiphenyl (0.90Kg, 1.89mol). Under the protection of nitrogen, the temperature is controlled at 70±5℃ to react for 4 hours. Cool down to 40±5°C, add ammonia water (68Kg), and stir for 8 hours. Cool down to 20±5°C and dilute with water (1110Kg). Dichloromethane extraction twice (244Kg, 170Kg). Combine the organic phases, wash sequentially with water and then with saturated brine. Add 3-mercaptopropyl ethyl sulfide-based silica (4.0Kg, used to remove heavy metal palladium) into the organic phase, and stir at 40±5°C for 20 hours. After filtration, the filtrate was concentrated under reduced pressure. The remainder was slurried sequentially with methyl tert-butyl ether and ethanol. Filter and dry in vacuo to obtain 4-(4-((7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-D]pyrimidin-2-yl)amino) -1H-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 34.6Kg, yield: 68.6%. LC-MS(ESI): m/z=539.3[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.46 (s, 1H), 8.94 (s, 1H), 7.76 (s, 1H), 7.38 (s, 1H), 7.33 to 7.35 (m, 1H) ,7.08-7.11(m,1H),6.91-6.95(m,1H),4.03-4.12(m,3H),3.73(s,3H),2.85-2.89(m,2H),2.39(s,3H) ,1.90-1.93(m,2H),1.55-1.60(m,2H),1.41(s,9H).
Comparative Example 1: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
Into a 100mL reaction flask, add 10% palladium on carbon (0.1g), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (2g, 9.2mmol), methanol (40mL) and N,N-Diisopropylethylamine (1.412 g, 10.9 mmol). Fill the bottle with hydrogen and control the hydrogen pressure at 0.5MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 21 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (2.1 g) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (2.2g) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 13.3g of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was precipitated, centrifuged, and the filter cake was vacuum dried to obtain 1.6 g of 2-chloro-6-methylthieno[3,2-D]pyrimidine as a product, with a yield of 54%. Methoxy substituted impurities in 20% yield.
Comparative Example 2: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
After replacing the solvent tetrahydrofuran in Example 2 with ethyl acetate, the solubility of 2-chloro-6-methylthieno[3,2-D]pyrimidine in ethyl acetate was poor, and only a small amount of product was formed, which was not calculated Specific yield.
Comparative example 3: 4-(p-toluenesulfonyl)-piperidine-1-tert-butyl carbonate (Compound G)
Triethylamine (25mL), N-BOC-4-hydroxypiperidine (5g) were added to a 100mL reaction flask. P-toluenesulfonyl chloride (7.1g) was added in batches while controlling the temperature at 10±10°C. After the addition, the temperature is controlled at 25±5℃ to react for 25 hours. Monitoring by LC-MS showed a large amount of unreacted raw materials and the reaction liquid was black and red.