














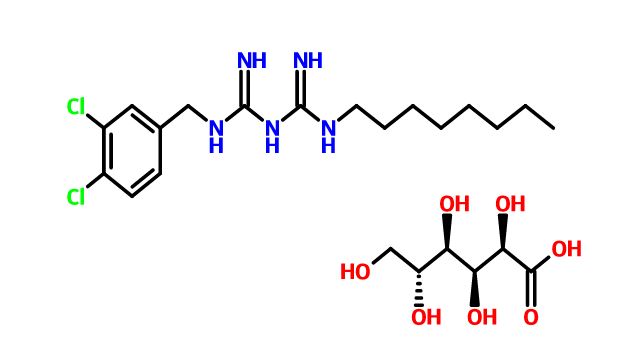
Olanexidine Gluconate
OPB-2045G, Gluconate olanexidin, Olanedine, OPB-2045, OPB 2045G,
(Olanedine®)Approved in Japan PMDA 2015-07-03, Olanedine® by Otsuka

A disinfectant used to prevent of postoperative bacterial infections.


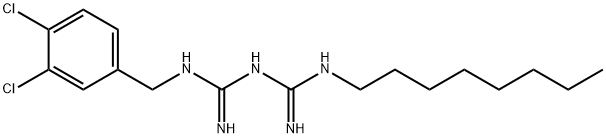
CAS .146510-36-3(Olanexidine free form),
Imidodicarbonimidic diamide, N-((3,4-dichlorophenyl)methyl)-N’-octyl
| C17H27Cl2N5 | |
| Formula Weight: | 372.341 |
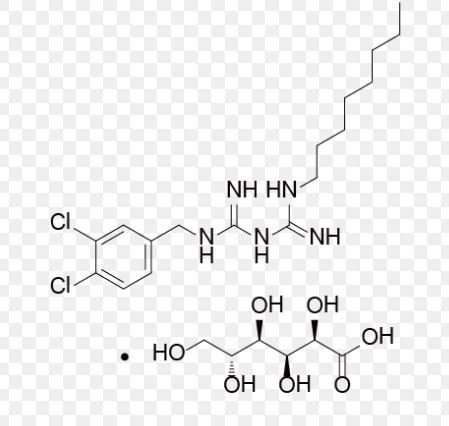
CAS 799787-53-4(Olanexidine Gluconate)
| 568.49 | |
| Formula | C17H27Cl2N5 ● C6H12O7 |
1-(3,4-Dichlorobenzyl)-5-octylbiguanide mono-D-gluconate
| オラネキシジングルコン酸塩 Olanexidine Gluconate  C17H27Cl2N5▪C6H12O7 : 568.49 [799787-53-4] |
Indication:Bacterial infection
Otsuka (Originator)

- Marketed Bacterial infections
Most Recent Events
- 16 Sep 2015 Launched for Bacterial infections (Prevention) in Japan (Topical)
- 03 Jul 2015 Registered for Bacterial infections (Prevention) in Japan (Topical) – First global approval
- 30 Sep 2014 Preregistration for Bacterial infections (Prevention) in Japan (Topical)

SEE ALSO
Olanexidine hydrochloride [USAN]
146509-94-6 HCL
RN: 218282-71-4 HCL HYDRATE
UNII: R296398ALN
Molecular Formula, C17-H27-Cl2-N5.Cl-H.1/2H2-O
Molecular Weight, 835.6192
Imidodicarbonimidic diamide, N-((3,4-dichlorophenyl)methyl)-N’-octyl-, monohydrochloride, hydrate (2:1)
INTRODUCTION
Olanexidine gluconate was approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Jul 03, 2015. It was developed and marketed as Olanedine® by Otsuka in Japan.
Olanexidine gluconate is an antiseptic/disinfectant compound with potent bactericidal activity against Gram-negative and Gram-positive bacteria, for use in preparing patients for surgery and preventing of postoperative bacterial infections.
Olanedine® is available as topical solution (1.5%), containing 3 g/200 mL, 0.15 g/10 mL and 0.375 g/25 mL, and the recommendation is applying appropriate amount of the drug.
PRODUCT PATENT
Kazuyoshi Miyata, Yasuhide Inoue, Akifumi Hagi, Motoya Kikuchi, Hitoshi Ohno, Kinji Hashimoto, Kinue Ohguro, Tetsuya Sato,Hidetsugu Tsubouchi, Hiroshi Ishikawa,Takashi Okamura, Koushi Iwata,
| Otsuka Pharmaceutical Co., Ltd., Otsuka Pharmaceutical Factory, Inc. |
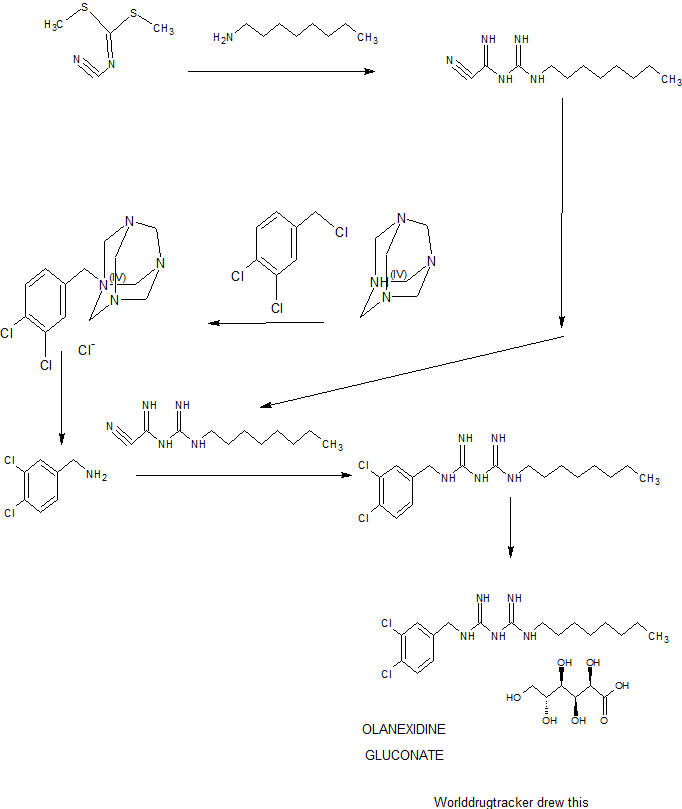
SYNTHESIS

PATENT
CN1065453A
http://www.google.com.au/patents/CN1065453A?cl=en
WO2008026757A1
https://google.com/patents/WO2008026757A1?cl=en
Example 1: l-cyano-3-n-octylguanidine
A 7.00-kg quantity of Compound (4) (54.16 mol) was dissolved in 105 liters of ethyl acetate, and the resulting mixture was cooled to 5°C or below. A 2.66-kg quantity of concentrated sulfuric acid (27.12 mol) was added thereto dropwise at a temperature of 4O0C or below while stirring. To the thus- obtained suspension of 1/2 sulfate of Compound (4) was added 5.06 kg of sodium dicyanamide (56.83 mol), and the resulting suspension was heated under reflux for 7 hours. The reaction solution was cooled to 400C or below, and 70 liters of water was added thereto. Subsequently, the resulting solution was heated to 80 to 900C (internal temperature) to distill the ethyl acetate off. The remaining liquid was cooled to 400C or below, and 70 liters of toluene was then added thereto, followed by the extraction of 1-cyano — 3-n-octyl guanidine at about 500C. The extracted toluene layer was washed with 35 liters of water at about 500C and cooled to 100C or below, followed by stirring for about 30 minutes. The resulting precipitated crystals were separated and washed with 7 liters of toluene. The resulting crystals were dried at 400C for 7.5 hours, yielding l-cyano-3-n- octylguanidine. 2007/067107
-16-
Yield: 9.11 kg (The yield was 85.7% based on the Compound(4).) White crystals having a melting point of 69 to 740C (no clear melting point was observed)
IR(KBr) spectrum: 3439, 3296, 2916, 2164, 1659, 1556, 1160, 718, and 572 cm“1
Thermogravimetric measurement/differential thermal analysis: 73.5°C (weak), an endothermic peak at 77.50C
1H-NMR(CDCl3) spectrum: 0.88 ppm (t, J = 6.6 Hz, 3H), 1.20-1.38 ppm (m, 10H), 1.43-1.62 ppm (m, 2H), 3.17 ppm (dd, J = 6.9 Hz, J = 6.0 Hz, 2H), 5.60-5.70 ppm (bs, 2H), 5.80-5.95 ppm (bs, IH)
Reference Example 2: Acidolysis of 1- (3,4-dichlorobenzyl) -5- octylbiguanide dihydrochloride
A 1-g quantity of 1- (3, 4-dichlorobenzyl) -5-octyl biguanide dihydrochloride was dissolved in 15 ml of 10% ethanol, followed by refluxing for 5 hours. HPLC analysis was conducted under the conditions described below.
The yield of 1-[N- (3,4-dichlorobenzyl) carbamoyl-3- octyl]guanidine (holding time: 9.84 minutes) was 0.91%, and the yield of 1- (N-octyl-carbamoyl) -3- (3, 4-dichlorobenzyl) guanidine
(holding time: 10.54 minutes) was 0.22%.
HPLC analysis conditions:
Column: YMC AM302 4.6 mm I. D. x 150 mm
Eluate: MeCN/0.05 M aqueous solution of sodium 1- octanesulfonate/acetic acid = 700/300/1
Detector: UV 254 nm
The physical property values of the resulting 1-[N- (3,4- dichlorobenzyl) carbamoyl-3-octyl] guanidine were as follows: NMR (DMSO-de) δ: 0.86 (3H, t, J = 6.0 Hz), 1.07-1.35 (1OH, m) , 1.35-1.49 (2H, m) , 2.95-3.15 (2H, m) , 4.12 (2H, d, J = 6.3 Hz), 6.78-7.40 (4H, m) , 7.23 (IH, dd, J = 2.1 Hz, J = 8.4 Hz), 7.46 (IH, d, J = 2.1 Hz), 7.54 (IH, d, J = 8.4 Hz)
The physical property values of the resulting 1- (N-octyl- carbamoyl) -3- (3, 4-dichlorobenzyl) guanidine were as follows: NMR (DMSO-d6) δ: 0.85 (3H, t, J = 6.6 Hz), 1.02-1.40 (12H, m) , 2.89-2.95 (2H, m) , 4.33 (2H, bs) , 5.76-7.00 (4H, m) , 7.28 (IH, dd, J = 2.1 Hz, J = 8.1 Hz), 7.52 (IH, d, J = 2.1 Hz), 7.58 (IH, d, J = 8.1 Hz)
Example 1: 1- (3, 4-dichlorobenzyl) -5-octylbiguanide monohydrochloride 1/2 hydrate
A 9.82-g quantity of Compound (2) (0.05 mol) and 10.63 g of 3, 4-dichlorobenzylamine (0.05 mol) were added to 49 ml of butyl acetate, followed by refluxing for 6 hours. The reaction solution was concentrated under reduced pressure, and a mixture of 12 ml of water and 47 ml of isopropyl alcohol was added and dissolved into the remainder. To the thus-obtained solution was added, dropwise, 10.13 g of concentrated hydrochloric acid. The resulting mixture was stirred at 28 to 300C for 30 minutes, and the precipitated crystals were then filtered out. The thus- obtained crystals were washed with a small amount of isopropyl alcohol, yielding 23.42 g of (non-dried) 1- (3, 4-dichlorobenzyl) – 5-octylbiguanide dihydrochloride. The resulting crystals were suspended in 167 ml of water without drying, the suspension was then stirred at 25 to 27°C for 2 hours, followed by separation of the crystals by filtration. The thus-obtained crystals were washed with a small amount of water and dried at 400C for 20 hours, yielding 17.05 g of 1- (3, 4-dichlorobenzyl) -5-octyl biguanide monohydrochloride 1/2 hydrate having a purity of 99.9% at a yield of 81.6%.
Example 2 : 1- (3, 4-dichlorobenzyl) -5-octylbiguanide dihydrochloride
A 100-g quantity of Compound (4) (0.774 mol) was dissolved in 1 liter of n-butyl acetate, and 37.6 g of concentrated sulfuric acid (0.383 mol) was added thereto while stirring. To the thus-obtained suspension of 1/2 sulfate of Compound (4) was added 68.9 g of sodium dicyanamide (0.774 mol), 7107
-18- and the resulting suspension was heated under reflux for 3 hours. The reaction solution was cooled to about 200C, and the organic layer thereof was sequentially washed with about 500 ml each of (i) 5% hydrochloric acid, (ii) 5% aqueous caustic soda solution, (iii) 5% aqueous sodium bicarbonate solution, and (iv) water.
To the thus-obtained n-butyl acetate solution of Compound (2) were added 118.5 g of Compound (3) (0.673 mol) and then 58.4 ml of concentrated hydrochloric acid while stirring. The reaction solution was heated, and about 800 ml of n-butyl acetate was distilled off under atmospheric pressure (ordinary pressure) , followed by heating the reaction solution under reflux for 3.5 hours . Subsequently, the reaction solution was cooled to about 400C, and 900 ml of isopropanol, 100 ml of water, and 134 ml of concentrated hydrochloric acid were added thereto. The mixture was stirred at 60 to 70°C for 1 hour and cooled to 100C or below and the precipitated crystals were then separated. The resulting crystals were washed with 200 ml of isopropanol and dried at 6O0C, yielding 1- (3, 4-dichlorobenzyl) -5-octylbiguanide dihydrochloride. Yield: 243.8 g (The yield was 81.3% based on the Compound (3).) Melting point: 228.90C IR(KBr) spectrum: 2920, 1682, 1634, 1337, 1035, 820, and 640 cm“1
PATENT
WO2004105745A1
PATENT
WO2009142715A1
PATENT
https://www.google.com/patents/US8334248
Olanexidine is a compound with high bactericidal activity having the chemical name 1-(3,4-dichlorobenzyl)-5-octylbiguanide. Research has been carried out into bactericides containing, olanexidine hydrochloride as an active ingredient (see Japanese Patent No. 2662343, etc.).
Olanexidine has very poor solubility in water, and hitherto known salts of olanexidine are also poorly soluble in water. For example, the solubility at 0° C. of olanexidine hydrochloride in water has been measured to be less than 0.05% (W/V), and the solubility of free olanexidine is a further order of magnitude less than this. Consequently, sufficient bactericidal activity cannot be expected of an aqueous solution merely having olanexidine dissolved therein, and moreover, depending on the conditions the olanexidine may precipitate out.
In the case of making an aqueous preparation of olanexidine in particular, to make the concentration of the olanexidine sufficient for exhibiting effective bactericidal activity, and to reduce the possibility of the olanexidine precipitating out, it has thus been considered necessary to use a dissolution aid such as a surfactant.
EXAMPLE 1 Preparation of an Aqueous Solution Aqueous Solution 1
20.9 g (50 mmol) of olanexidine hydrochloride hemihydrate was added to 250 mL of a 1 N aqueous sodium hydroxide solution, and the suspension was stirred for 1.5 hours at room temperature (25° C.). The solid was filtered off, and washed with water. The solid obtained was further suspended in 250 mL of purified water, the suspension was stirred for 5 minutes at room temperature, and the solid was filtered off, and washed with water. This operation was carried out once more to remove sodium chloride formed. The solid obtained (free olanexidine) was put into purified water in which 8.9 g (50 mmol) of gluconolactone had been dissolved, and the mixture was stirred at room temperature until the solid dissolved, and then purified water was further added to give a total volume of 300 mL. The concentration of olanexidine in the aqueous solution obtained was measured by using high performance liquid chromatography to be 6% in terms of free olanexidine.
This aqueous solution was still transparent and colorless even after being left for several months at room temperature.
CLIP
http://dmd.aspetjournals.org/content/28/12/1417/F9.expansion.html
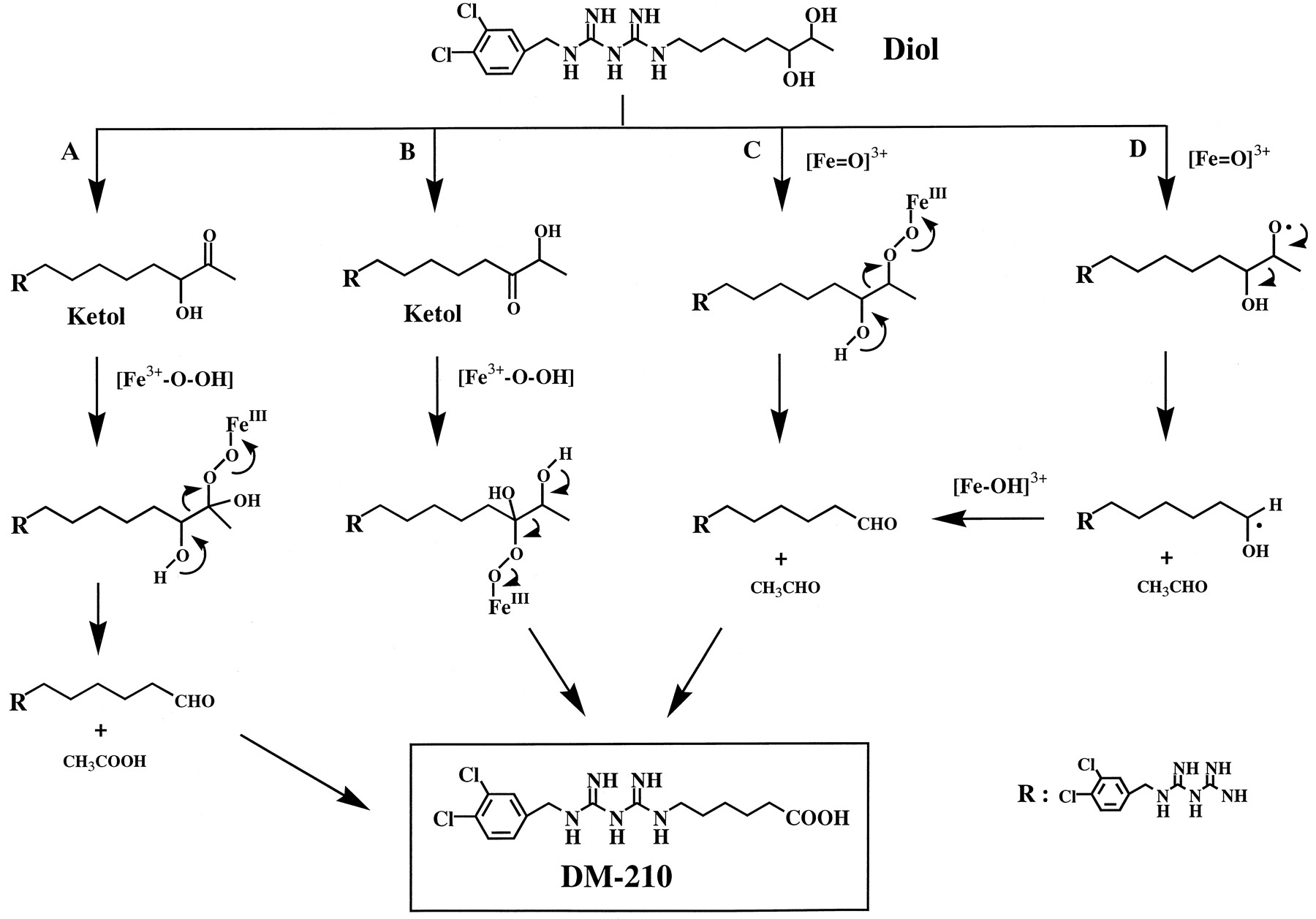
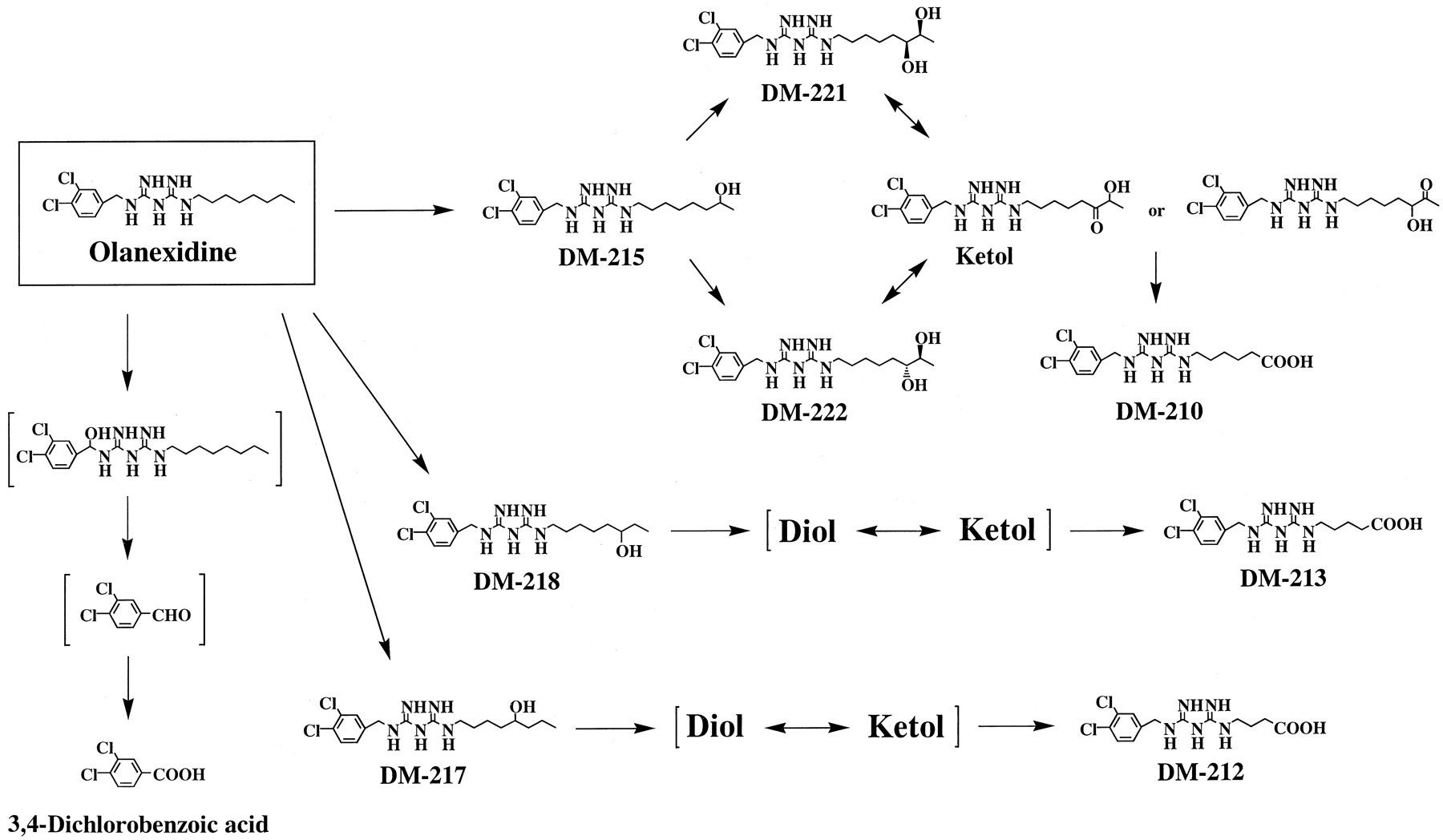

REFERENCES
http://www.otsukakj.jp/en/news/photo/photo-14423716650.pdf
| Patent ID | Date | Patent Title |
|---|---|---|
| US8979785 | 2015-03-17 | Fluid application device and method |
| US8911771 | 2014-12-16 | Fluid application device and method |
| US8858484 | 2014-10-14 | Fluid application device and method |
| US2013330114 | 2013-12-12 | FLUID APPLICATION DEVICE AND METHOD |
| US2012095254 | 2012-04-19 | METHOD AND APPARATUS FOR PREPARING A SOLUTION OF A SHEAR SENSITIVE MATERIAL |
| US7868207 | 2011-01-11 | PROCESS FOR PRODUCING 1-(3, 4-DICHLOROBENZYL)-5-OCTYLBIGUANIDE OR A SALT THEREOF |
| US2010331421 | 2010-12-30 | DISINFECTANT AND/OR BACTERICIDAL AQUEOUS COMPOSITIONS |
| US2010331423 | 2010-12-30 | AQUEOUS SOLUTION OF OLANEXIDINE, METHOD OF PREPARING THE AQUEOUS SOLUTION, AND DISINFECTANT |
| US7829518 | 2010-11-09 | Aqueous solution of olanexidine, method of preparing the aqueous solution, and disinfectant |
| US7825080 | 2010-11-02 | Aqueous solution of olanexidine, method of preparing the aqueous solution, and disinfectant |
| Patent ID | Date | Patent Title |
|---|---|---|
| US7622469 | 2009-11-24 | 2, 4-diamino-1, 3, 5-triazine derivatives |
| US2009287021 | 2009-11-19 | METHOD AND APPARATUS FOR PREPARING A SOLUTION OF A SHEAR SENSITIVE MATERIAL |
| US2007053942 | 2007-03-08 | Disinfectant and/or bactericidal aqueous compositions |
| EP0507317 | 1997-01-15 | BIGUANIDE DERIVATIVES, MANUFACTURING METHOD THEREOF, AND DISINFECTANTS CONTAINING THE DERIVATIVES |
| EP0507317A2 * | Apr 3, 1992 | Oct 7, 1992 | Otsuka Pharmaceutical Co., Ltd. | Biguanide derivatives, manufacturing method thereof, and disinfectants containing the derivatives |
| EP1634589A1 * | May 25, 2004 | Mar 15, 2006 | Otsuka Pharmaceutical Co., Ltd. | Aqueous olanexidine solution, method of preparing the same, and disinfectant |
| Reference | ||
|---|---|---|
| 1 | * | TSUBOUCHI H ET AL: “Synthesis and Structure-Activity Relationships of Novel Antiseptics” BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, OXFORD, GB, vol. 7, no. 13, 8 July 1997 (1997-07-08), pages 1721-1724, XP004136287 ISSN: 0960-894X |
//////////Olanexidine Gluconate, OPB-2045G, (Olanedine®, Approved, japan 2015-07-03, Olanedine, Otsuka, PMDA, Olanexidine, オラネキシジングルコン酸塩 , Gluconate olanexidin, Olanedine, OPB-2045, OPB 2045G, JAPAN 2015
CCCCCCCCN=C(N)NC(=NCC1=CC(=C(C=C1)Cl)Cl)N
Clc1ccc(CNC(=N)NC(=N)NCCCCCCCC)cc1Cl.O=C(O)[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO