














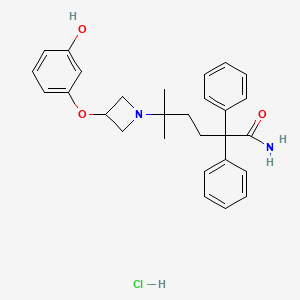

PF-3635659
CAS 931409-24-4 FREE FORM
Molecular Formula, C28-H32-N2-O3, Molecular Weight, 444.5718
1-Azetidinepentanamide, 3-(3-hydroxyphenoxy)-delta,delta-dimethyl-alpha,alpha-diphenyl-
5-[3-(3-hydroxyphenoxy)azetidin-1-yl]-5-methyl-2,2-diphenylhexanamide;hydrochloride
| Molecular Formula | C28H33ClN2O3 |
|---|---|
| Synonyms |
PF-3635659 (hydrochloride) 1079781-31-9 5-[3-(3-Hydroxy-phenoxy)-azetidin-1-yl]-5-methyl-2,2-diphenyl-hexanoic acid amide hydrochloride |
| Molecular Weight | 481 g/mol |
READ
Pf03635659 has been used in trials studying the treatment of Chronic Obstructive Pulmonary Disease.
|
~65%
931409-24-4 |
| Literature: PFIZER LIMITED Patent: WO2008/135819 A1, 2008 ; Location in patent: Page/Page column 14; 15 ; WO 2008/135819 A1 |
|
~%
931409-24-4 |
| Literature: Dillon, Barry R.; Roberts, Dannielle F.; Entwistle, David A.; Glossop, Paul A.; Knight, Craig J.; Laity, Daniel A.; James, Kim; Praquin, Celine F.; Strang, Ross S.; Watson, Christine A. L. Organic Process Research and Development, 2012 , vol. 16, # 2 p. 195 – 203 |
|
~%
931409-24-4 |
| Literature: Glossop, Paul A.; Watson, Christine A. L.; Price, David A.; Bunnage, Mark E.; Middleton, Donald S.; Wood, Anthony; James, Kim; Roberts, Dannielle; Strang, Ross S.; Yeadon, Michael; Perros-Huguet, Christelle; Clarke, Nicholas P.; Trevethick, Michael A.; MacHin, Ian; Stuart, Emilio F.; Evans, Steven M.; Harrison, Anthony C.; Fairman, David A.; Agoram, Balaji; Burrows, Jane L.; Feeder, Neil; Fulton, Craig K.; Dillon, Barry R.; Entwistle, David A.; Spence, Fiona J. Journal of Medicinal Chemistry, 2011 , vol. 54, # 19 p. 6888 – 6904 |
![5-[3-(3-hydroxyphenoxy)azetidin-1-yl]-5-methyl-2,2-diphenylhexanenitrile structure](https://www.chemsrc.com/caspic/250/931409-66-4.png)
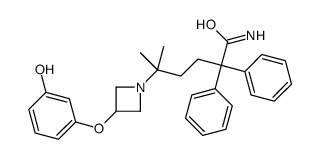

PAPER
Organic Process Research & Development (2012), 16(2), 195-203.
https://pubs.acs.org/doi/10.1021/op200233r

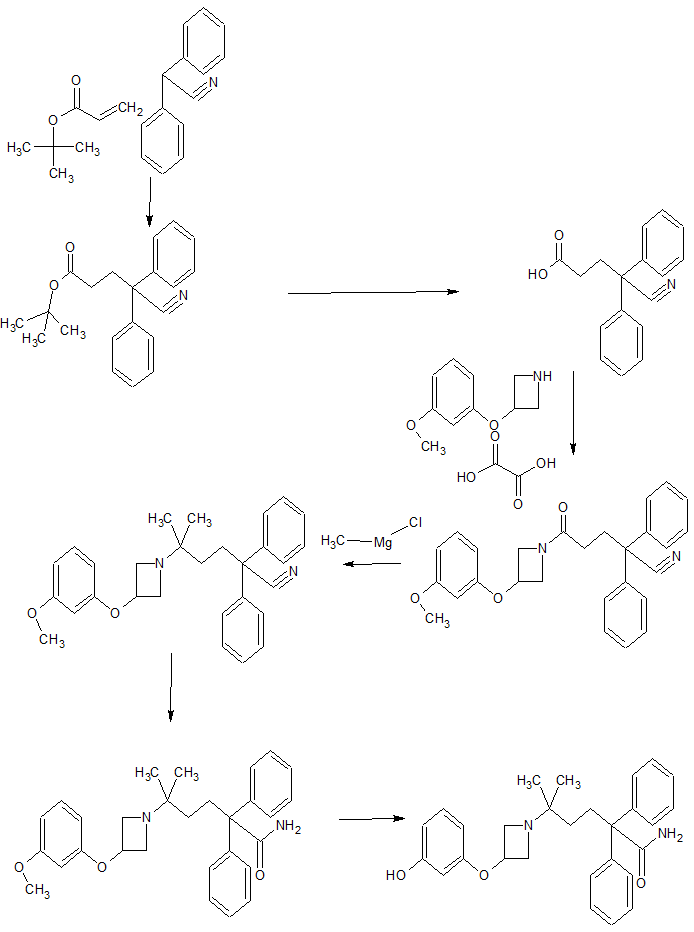
An efficient and scalable process for the synthesis of muscarinic antagonist, PF-3635659 1, is described, illustrating redesign of an analogue-targeted synthesis which contained a scale-limiting rhodium-activated C–H amination step. The final route includes a reproducible modified Bouveault reaction which has not previously been reported on a substrate of this complexity, or on such a scale with over 5 kg of the requisite gem-dimethylamine prepared via this methodology.
5-[3-(3-Hydroxyphenoxy)azetidin-1-yl]-5-methyl-2,2-diphenylhexanamide (1).
First Discovery Route.
To a solution of 5-methyl-2,2-diphenyl-5-{3-[3-(prop-2-en-1-yloxy)phenoxy]azetidin1-yl}hexane nitrile 9 (2.8 g, 6.01 mmol) in 3-methyl-pentan-3-ol (30 mL) was added potassium hydroxide (6.7 g, 120 mmol) and the resulting solution was stirred at 120 ºC for 22 hours. The reaction was cooled to room temperature and concentrated in vacuo. The residue was partitioned between ethyl acetate (100 mL) and water (50 mL). The aqueous layer was re-extracted with ethyl acetate (2 x 50 mL). The combined organic layers were dried with MgSO4 and concentrated in vacuo to yield 5-methyl-2,2-diphenyl-5-(3-{3- (propenyl)oxy-phenoxy}-azetidin-1-yl)-hexanamide 10 as a yellow oil (3 g, 6.01 mmol, 100%) which was taken on crude to the next step. To a solution of 5-methyl-2,2-diphenyl-5-(3-{3-(propenyl)oxy-phenoxy}-azetidin-1-yl)- hexanoic acid amide 10 (3.0 g, 6.01 mmol) in methanol (100 mL) was added a 2M aqueous hydrochloric acid solution (30 mL, 15 mmol) and the resulting solution was stirred at 60 ºC for 40 minutes. The volatile solvents were removed in vacuo and the remaining aqueous residue was basified with a saturated aqueous sodium hydrogen carbonate solution. The aqueous layer was extracted with ethyl acetate (3 x 100 mL) and the combined organic layers were dried with magnesium sulphate and concentrated in vacuo.
The crude residue was purified by flash chromatography eluting in ethyl acetate:methanol:ammonia (90:10:1) / pentane (50/50) to yield the title compound 1 as a colourless foam (1.5 g, 3.37 mmol, 54.5%).
Second Discovery Route.
To a solution of 5-[3-(3-methoxyphenoxy)azetidin-1-yl]-5-methyl-2,2-diphenylhexanamide 19 (9.0 g, 19.6 mmol) in dichloromethane (1.25 L) at 0 ºC was dropwise added a solution of boron tribromide (1M in dichloromethane, 58.9 mL, 58.9 mmol) and the mixture stirred for 2 hours at 0 ºC to 20 oC. The mixture was cooled to 0 ºC and quenched with 1M aqueous sodium hydroxide solution (200 mL). The reaction mixture was allowed to warm to 20 oC and stirred as such for 1 hour. The layers were separated and the aqueous layer was extracted with ethyl acetate (2 x 200 mL). The combined organic layers were dried with sodium sulphate and concentrated in vacuo. The crude residue was purified by column chromatography eluting in ethyl acetate:methanol:ammonia (90:10:1) / pentane (50/50) to yield the title compound 1 as a white foam (3.4 g, 7.64 mmol, 39%)
1H NMR (MeOD): δ=0.93 (s, 6H), 1.09-1.14 (m, 2H), 2.38-2.42 (m, 2H), 3.11-3.15 (m, 2H), 3.43-3.47 (m, 2H), 4.57-4.62 (m, 1H), 6.19-6.23 (m, 2H), 6.36 (d, 1H), 7.02 (t, 1H), 7.23-7.38 (m, 10H); MS: m/z 445 [M+H]+.
PAPER
Journal of Medicinal Chemistry (2011), 54(19), 6888-6904.
https://pubs.acs.org/doi/10.1021/jm200884j

A novel tertiary amine series of potent muscarinic M3 receptor antagonists are described that exhibit potential as inhaled long-acting bronchodilators for the treatment of chronic obstructive pulmonary disease. Geminal dimethyl functionality present in this series of compounds confers very long dissociative half-life (slow off-rate) from the M3 receptor that mediates very long-lasting smooth muscle relaxation in guinea pig tracheal strips. Optimization of pharmacokinetic properties was achieved by combining rapid oxidative clearance with targeted introduction of a phenolic moiety to secure rapid glucuronidation. Together, these attributes minimize systemic exposure following inhalation, mitigate potential drug–drug interactions, and reduce systemically mediated adverse events. Compound 47 (PF-3635659) is identified as a Phase II clinical candidate from this series with in vivo duration of action studies confirming its potential for once-daily use in humans.
Patent
WO-2020261160
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020261160
Methods and intermediates for preparing the hydrochloride salt of PF-3635659 ,
Cholinergic muscarinic receptors are members of the G-protein coupled receptor super-family and are further divided into 5 subtypes, M to Ms. Muscarinic receptor sub-types are widely and differentially expressed in the body. Genes have been cloned for all 5 sub-types and of these, Mi, M>, and Ms receptors have been extensively pharmacologically characterized in animal and human tissue. Mi receptors are expressed in the brain (cortex and hippocampus), glands and in the ganglia of sympathetic and parasympathetic nerves. M2 receptors are expressed in the heart, hindbrain, smooth muscle and in the synapses of the autonomi c nervous system. Ms receptors are expressed m the brain, glands and smooth muscle. In the airways, stimulation of Ms receptors evokes contraction of airway smooth muscle leading to bronchoeonstnction, while in the salivary-gland Ms receptor stimulation increases fluid and mucus secretion leading to increased salivation. M2 receptors expressed on smooth muscle are understood to be pro-contractile while pre-synaptic M2 receptors modulate acetylcholine release from parasympathetic nerves. Stimulation of M2 receptors expressed in the heart produces bradycardia.
[0003] Short and long-acting muscarinic antagonists are used in the management of asthma and chronic obstructive pulmonary disease (COPD); these include the short acting agents Atrovent® (ipratropium bromide) and Oxivent® (oxitropium bromide) and the long acting agent Spiriva® (tiotropium bromide). These compounds produce bronchodilation following inhaled administration. In addition to improvements in spirometric values, anti-muscarinic use in COPD is associated with improvements m health status and quality of life scores. As a consequence of the wide distribution of muscarinic receptors in the body, significant systemic exposure to muscarinic antagonists is associated with effects such as dry mouth, constipation, mydriasis, urinary retention (all predominantly mediated via blockade of M3 receptors) and tachycardia (mediated by blockade of M2 receptors).
[0004] A newer M3 receptor antagonist that is in the carboxamide family is 5-[3-(3-hydroxyphenoxy)azetidin-l-yl]-5-methyl-2,2-diphenylhexanamide hydrochloride. This carboxamide compound exhibits the following structure (formula II):
[0005] To date, it has not been appreciated that 5-[3-(3-hydroxyphenoxy)azetidin-l-yl]-5-methyl-2,2-diphenylhexanamide hydrochloride can be synthesized from the benzoate salt of 5-[3-(3-hydroxyphenoxy)azetidin~l~y!]-5-methyl-2,2-diphenylhexanenitrile Therefore, there is a need for methods and intermediates used to efficiently prepare 5-[3-(3-hydroxyphenoxy)azetidin~l~y!]-5-methyl-2,2-diphenylhexanamide hydrochloride of good quality from the benzoate salt of 5~[3~ (3~hydroxyphenoxy)azetidin-l-yl]-5-rn ethyl-2, 2-diphenylhexanenitrile.
Reaction Scheme 1 -Preparation of Crude Carboxamide Hydrochloride
formula I formula II
[0061] The coupled benzoate compound of formula 1 can be reacted with KOH, 2-methyl-2-butano!, water, then HC1 aqueous, HC1, and TBME to obtain the crude carboxamide hydrochloride of formula II. The benzoate salt of the nitrile provides for easier purification of the nitrile.
[0062] The reagents useful in the preparation of 5-[3-(3-hydroxyphenoxy)azetidin-l-yl]-5-metiiyl-2,2-diphenyl-hexanamide hydrochloride include a base and an alcohol In some embodiments, a useful base includes potassium hydroxide, while a useful alcohol includes tertiary amyl alcohol also known as 2-methyl-2-butanol. The reaction of the benzoate compound of formula II in tertiary amyl alcohol and potassium hydroxide can be carried in a temperature range from about 85 ± 5°C to about 103 ± 2°C. In a later stage, the temperature of 103 ± 2°C can be maintained in that range for from about 30 hours to about 65 hours. A cooling period to about room temperature is followed by adjusting the pH to a range from about 6.5 to about 8.0. Hydrochloric acid is added to the product of this initial reaction to form a crude carboxamide hydrochloride compound of formula II. The initially isolated crude carboxamide hydrochloride compound of formula II can be washed with an alcohol and then washed with, or slurried in an ether. In some embodiments, the alcohol can be tertiary amyl alcohol and the ether can be methyl tertiary butyl ether.
[0063] In various embodiments, the crude 5-[3-(3-hydroxyphenoxy)azetidin-l-yl]-5-methyl-2,2-diphenylhexanamide hydrochloride can be further purified by treating this carboxamide hydrochloride compound with a slurry of activated charcoal, for example, commercially available ENQPC, PF133 or PF511 SPL (A) carbon, in isopropyl alcohol and water at 85 ± 5°C and filtering as illustrated m the Reaction Scheme 2 below:
Reaction Scheme 2 – Purification of Carboxamide Hydrochloride
Reaction Scheme 3 – Preparation of the Coupled Compound Benzoate
O
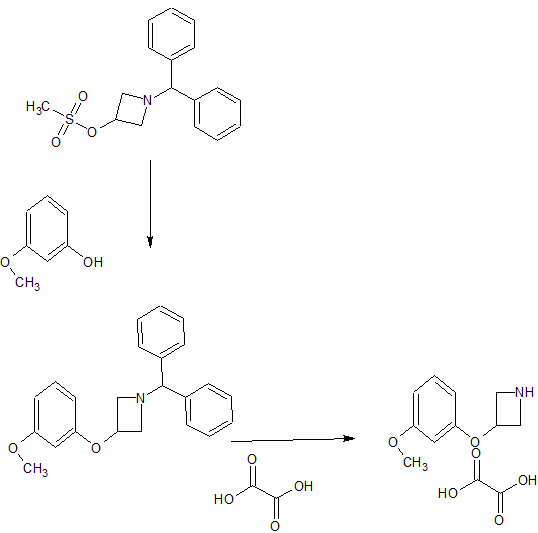
[0065] In some embodiments, the benzyl coupled compound of formula III is prepared by reacting an azetidine mesyl HC1 1 -(5-cyano-2-methyl-5,5-diphenylpentan-2-yl)azetidin-3-yl methanes ulfonate hydrochloride with a reagent comprising benzyl resorcinol as illustrated in the Reaction Scheme 4 below:
Reaction Scheme 4 – Preparation of the Benzyl Coupled Compound
In Reaction Scheme 4, the azetidine mesyl hydrochloride of formula IV
is reacted with benzyl resorcinol of formula V
The reagent can comprise benzyl resorcinol and, in some aspects, acetonitrile, a carbonate salt of either cesium or potassium, sodium hydroxide, water, ethyl acetate, hexanes or a mixture thereof. The order of addition of reagents in this step overcomes the need for specific equipment (e.g., a bespoke/unusual agitator) and allows the step to be run in a general purpose reactor.
[0066] Benzyl resorcinol is commercially available and can be obtained commercially, for example, from Sigma Aldrich Corp. In various embodiments, benzyl resorcinol of formula V can be prepared by reacting resorcinol with benzyl chloride to form benzyl resorcinol according to the Reaction Scheme 5 below:
Reaction Scheme 5 — Preparation of Benzyl Resorcinol
Resorcinol DMF/Hexane
Toluene Benzyl Resorcinol
or
3-{benzyioxy) phenol
V
[0067] In certain aspects, the benzyl resorcinol is prepared by reacting resorcinol with benzyl chloride m a reagent which can include potassium carbonate, dimethylformamide, water, sodium hydroxide, toluene, hydrochloric acid, hexanes or a combination thereof. In some instances, benzyl resorcinol seeding material may also be added. For the conversion of the resorcinol to the benzyl resorcinol (V), the developed chemistry’- allows effective removal of remaining resorcinol starting material and dibenzyl impurity to give the benzyl resorcinol product in good yield and quality.
Reaction Scheme 6 – Preparation of Azetidine Mesyl Hydrochloride
Azetidine alcohol Azetidine mesyl
VI hydrochloride
Reaction Scheme 7 – Preparation of Azetidine Alcohol
Scheme 8 – Preparation of Diphenyl Amine
Reaction Scheme 9 Preparation of Diphenyl Chloro Amide
Reaction Scheme 10 – Preparation of Diphenyl Alkene
3-methyl-3-buien-t-ol Mesyi Alkene Diphenyl Alkene
PATENT
WO2007034325
https://patents.google.com/patent/WO2007034325A1/en
The compound was originally claimed without an action as example 108 in WO2007034325 , for the treatment of chronic obstructive pulmonary disease, and this is the first filing from Pfizer relating to the compound since the program was presumed discontinued in 2011.
Example 108 5-r3-(3-Hvdroxyphenoxy)azetidin-1-vπ-5-methyl-2,2-diphenylhexanamide

Boron tribromide (1M in dichloromethane, 1.75mL, 1.75mmol) was added to an ice-cooled solution of the product of example 100 (200mg, 0.44mmol) in dichloromethane (5mL) and the mixture was stirred at O0C for 1 hour. Further boron tribromide (1M in dichloromethane, 0.5mL, O.δmmol) was added and the mixture was stirred at O0C for 30 minutes. The reaction was then quenched with 1M sodium hydroxide solution (5mL), diluted with dichloromethane (2OmL) and stirred at room temperature for 40 minutes. The aqueous layer was separated, extracted with ethyl acetate (2x25mL) and the combined organic solution was dried over magnesium sulfate and concentrated in vacuo. Purification of the residue by column chromatography on silica gel, eluting with pentane:ethyl acetate/methanol/0.88 ammonia (90/10/1), 75:25 to 50:50, afforded the title compound as a colourless foam in 91% yield, 176mg.
1HNMR(400MHz, CDCI3) δ: 1.10(s, 6H), 1.22-1.34(m, 2H), 2.42-2.55(m, 2H), 3.28-3.40(m, 2H), 3.65-3.88(m, 2H), 4.70-4.80(m, 1H), 5.55-5.70(brs, 2H), 6.23-6.36(m, 2H), 6.45-6.53(m, 1H), 7.03-7.12(m, 1H), 7.19-7.39(m, 10H); LRMS ESI m/z 445 [M+H]+ E
PATENT
WO2018167804
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018167804
It does however, follow on from WO2018167804 , assigned solely to Mylan , claiming amorphous and crystalline forms designated as Forms I-XI, for treating allergy, and this seems to confirm the potential of the candidate is being revisited, and possibly licensed.
(5-[3-(3-Hydroxyphenoxy)azetidin-l-yl]-5-methyl-2,2-diphenylhexanamide hydrochloride has a structure depicted below as Compound-A.
Compound-A
Compound-A is a muscarinic antagonist useful for treating allergy or respiratory chronic obstructive pulmonary disease.
Compound-A and pharmaceutically acceptable salts are claimed in U.S. Pat. No. 7,772,223 B2 and one of its non-solvated crystalline forms is claimed in U.S. Pat. No. 8,263,583 B2.
Examples:
Example 1: Processes for the preparation of amorphous form of Compound-A.
Compound-A (5 g) was dissolved in methanol (150 ml) at 60-65°C. The solution was filtered at 60-65°C to remove undissolved particulate and then cooled to 25-30°C. The clear solution of Compound-A was subjected to spray drying in a laboratory Spray Dryer (Model Buchi-290) with a 5 ml/min feed rate of the solution and inlet temperature at 75°C with 100% aspiration to yield an amorphous form of Compound-A.
///////////// PF-3635659, PF 3635659
CC(C)(CCC(C1=CC=CC=C1)(C2=CC=CC=C2)C(=O)N)N3CC(C3)OC4=CC=CC(=C4)O.Cl