















Pirlindole
- Molecular FormulaC15H18N2
- Average mass226.317 Da
Pirlindole (Lifril, Pyrazidol) is a reversible inhibitor of monoamine oxidase A (RIMA) which was developed and is used in Russia as an antidepressant.[1]:337 It is structurally and pharmacologically related to metralindole.
Biovista is investigating BVA-201, a repurposed oral formulation of pirlindole mesylate, for the potential treatment of multiple sclerosis
SYN 1
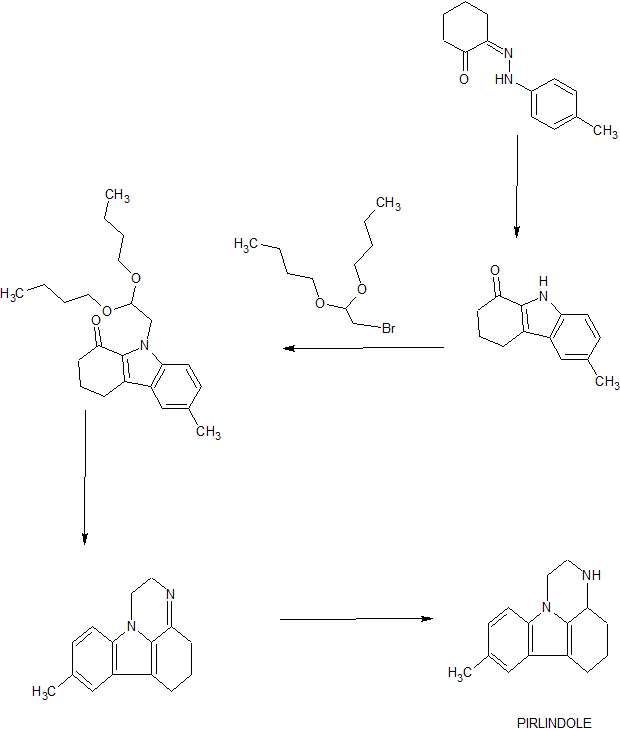
SYN 2
PAPER
Khimiko-Farmatsevticheskii Zhurnal (1986), 20(3), 300-3.
PATENT
U.S.S.R. (1986), SU 276060
PAPER
Sudebno-meditsinskaia ekspertiza (1989), 32(4), 49-50
PAPER
Journal of Pharmaceutical and Biomedical Analysis
https://www.sciencedirect.com/science/article/abs/pii/S0731708598002131
PATENT
claiming method for resolving racemic mixture of pirlindole hydrochloride into enantiomerically pure (S)-pirlindole and/or (R)-pirlindole,
Pirlindole, 2, 3, 3a, 4, 5, 6-hexahydro-lH-8-methyl-pyrazine
[3, 2, 1-j , k] carbazole, is a tetracyclic compound of the formula I
(I)
Pirlindole is a reversible monoamine oxidase A inhibitor being up to date useful as a medicament in the treatment of depression.
Pirlindole has an asymmetric carbon atom which implies that there are two enantiomers, (S) -pirlindole and (R) -pirlindole .
The state of the art teaches several methods for the enantiomeric separation of pirlindole. For example, The Journal of Pharmaceutical and Biomedical Analysis, 18(1998) 605- 614, “Enantiomeric separation of pirlindole by liquid chromatography using different types of chiral stationary phases”, Ceccato et al, discloses the enantiomeric separation of pirlindole by liquid chromatography (LC) using three different chiral stationary phases.
Further, The Journal of Pharmaceutical and Biomedical Analysis 27(2002) 447-455, “Automated determination of pirlindole enantiomers in plasma by on-line coupling of a pre-column packed with restricted access material to a chiral liquid chromatographic column”, Chiap et al., discloses the use of a pre-column packed with restricted access material for sample clean up coupled to a column containing a cellulose based chiral stationary phase for separation and quantitative analysis of the enantiomers .
According to the prior art, Chirality 11:261-266 (1999) all attempts to obtain the enantiomers of pirlindole by selective crystallization with optically active acids failed, and it was only possible to obtain at laboratory scale (few grams) as hydrochloride salt, using derivatization technique in conjunction with preparative chromatography.
The characteristics of the process disclosed in the state of the art limit in a definitive way, its implementation on an industrial or semi-industrial scale due to the necessity to use a separation by chromatography on a large scale which makes the process very costly, difficult to implement and with poor reproducibility. .
EXAMPLE 7
(R) -Pirlindole mesylate
Starting from 10 g of (R) -pirlindole (S) -mandelate obtained in Example 1 and following the procedure described in Example 5 using methanesulfonic acid as pharmaceutical acceptable acid, ,
7.4 g (0.023 mole) of (R) -pirlindole mesylate were obtained (yield = 85.2% ). Chiral HPLC (enantiomeric purity = 98.0%).
XAMPLE 9
(S) -pirlindole mesylate
Starting from 10 g of (S) -pirlindole (R) -mandelate obtained in Example 2 and following the procedure described in Example 6 using methanesulfonic acid as pharmaceutical acceptable acid, 6.8 g (0.021 mole) of (S) -pirlindole mesylate were obtained (yield = 77.8%). Chiral HPLC (enantiomeric purity = 98.0%).
PATENT
Process for the preparation of pirlindole . useful for treating depression.
Pirlindole (8-methyl-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole) of formula I
Compound Formula I
also described as Pyrazidole™ represents a new class of original tetracyclic antidepressants, the pyrazinocarbazole derivatives. The drug was synthesized and characterized at the end of the 1960s and was marketed as an anti-depressant in 1975. Current clinical trials have demonstrated to be a highly effective short-acting and safe drug.
[0003] Pirlindole is a selective, reversible inhibitor of MAO-A. In-vitro evidence suggest the catalytic oxidation of Pirlindole into dehydro-pirlindole by MAO-A. Dehydro-pirlindole may be a more potent slowly reversible inhibitor of MAO-A and this might explain the persistence of MAO-A inhibition in-vivo (MAO-The mother of all amine oxidases, John P.M. Finberg et al. 1998, Springer).
[0004] Pirlindole chemical structure is composed of one stereogenic centre which indicates the existence of two enantiomers, the ( ?)-Pirlindole and the (S)-Pirlindole.
[0005] Although Pirlindole pharmacological data and the clinical use were performed on the racemate, recently there have been increasing interest in the pharmacological profile of each enantiomer (WO 2015/171005 Al).
[0006] International patent publication WO 2015/171003A1 filed 9th May 2014 discloses a resolution of racemic pirlindole into optically active pirlindole. The Resolution-Racemization-Recycle (RRR) synthesis described involves derivatization by preparation of pairs of diastereomers in the form of salts from an optically active organic acid. These diastereomers can be separated by conventional techniques such as crystallisation. Although it is a very efficient procedure to prepare laboratorial scale or pre-clinical batch of (/?)- or (S)-Pirlindole, it is not economically convenient at an industrial scale because the process relies on Pirlindole racemate as the starting material.
[0007] Andreeva et al. (Pharmaceutical Chemistry 1992, 26., 365-369) discloses the first isolation of Pirlindole enantiomers in isolated form. ( ?)-Pirlindole of formula II
was isolated as an hydrochloride salt from a racemic base by the fractional crystallization of racemic pirlindole salt with (+)-camphor-10-sulfonic acid. (S)-Pirlindole formula III
was also isolated as an hydrochloride salt although via asymmetric synthesis from the 6-methyl-2,3,4,9-tetrahydro-lH-carbazol-l-one IV
[0008] Compound of formula IV was reacted with chiral auxiliary (S)-(-)-a-methylbenzylamine to afford asymmetric (S)-6-methyl-N-(l-phenylethyl)-2,3,4,9-tetrahydro-lH-carbazol-l-imine V
[0009] Compound of formula V was subjected to stereoselective reduction with sodium borohydride in ethanol. According to Andreeva et al. the reaction might occur through directed intramolecular hydride transfer after formation of a complex between compound of formula V and reducing agent to afford (S)-6-methyl-N-((S)-l-phenylethyl)-2,3,4,9-tetrahydro-lH-carbazol-l-amine VI
[0010] Compound of formula VI is reacted with ethylene glycol ditosylate by ethylene bridge formation under alkaline conditions to yield (S)-8-methyl-3-((S)-l-phenylethyl)-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole VII.
[0011] Alkaline agent is sodium hydride (NaH), in the presence of dimethyl sulfoxide (DMSO) or dimethylformamide (DMF).
[0012] The ratio between alkaline agent, compound of formula VI and ethylene glycol ditosylate is 1.2:1:1.
[0013] The cyclization reaction occurs at room temperature for a period of 4.5 hours. [0014] Compound of formula VII was subjected to catalytic hydrogenolysis conditions to afford the desired hydrochloride salt of compound of formula III.
[0015] The hydrogenolysis reaction was catalysed by Palladium on charcoal (Pd content 0.1 g, 9 mol%) and was conducted in methanol. The conversion of compound of formula VII into compound of formula III was performed under a hydrogen pressure of 1.8-2.0 MPa at 22 °C for a period of 17h.
[0016] The work-up conditions for the hydrogenolysis reaction involved neutralization with ammonia solution followed by benzene recrystallization. The hydrochloride salt of compound of formula III was formed from addition of hydrochloric acid to a solution of free base in ethanol.
[0017] The process yielded (S)-Pirlindole hydrochloride with a final yield of 10% with respect to the intermediate VI.
[0018] The mixture of sodium hydride with DMSO generates dimsyl anion. This anion is very often used in laboratory scale, but because it is unstable its use on large scale should be under specific precautions. Dimsyl anion decomposition is exotermic. It is reported that dimsyl anion decomposition starts even at 20 °C, and above 40 °C it decomposes at an appreciable rate (Lyness, W. I. et ai, U.S. 3,288,860 1966, CI. 260-607).
[0019] The mixture of DMF and sodium hydride is reported in ‘Sax & Lewis’s Dangerous Properties of Industrial Materials’ to give a violent reaction with ignition above 50 °C. Buckey, J. et ai, Chem. Eng. News 1982, 60(28), 5, describes the thermal runaway of a pilot plant reactor containing sodium hydride and DMF from 50 °C. Accelerated Rate Calorimetry (ARC) tests showed exothermic activity as low as 26 °C. Similar behaviour was also seen with DMA. De Wall, G. et ai, Chem. Eng. News 1982, 60(37), 5, reports a similar incident, wherein runaway started at 40 °C, and rose 100 °C in less than 10 minutes, boiling off most of the DMF.
[0020] There exists a need for safe, industrial- and eco-friendly processes for the preparation of Pirlindole enantiomers. These facts are disclosed in order to illustrate the technical problem addressed by the present disclosure.
[0068] In an embodiment, the preparation of (S)-8-methyl-3-((S)-l-phenylethyl)-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole, compound of formula VII was carried out as follow.
[0069] In an embodiment, in a 2 L three necked round bottomed flask equipped with magnetic stirrer, ethylene glycol ditosylate (73 g, 197 mmol) and DMI (240 mL) were loaded. To the resulting clear solution, NaH (60% suspension in mineral oil, 15.8 g, 394 mmol) was added carefully. To the resulting suspension a solution of VI ((S)-6-methyl-N-((S)-l-phenylethyl)-2,3,4,9-tetrahydro-lH-carbazol-l-amine) (30 g, 98.5 mmol) in DMI (60 mL) was added dropwise at 60 °C. The mixture was stirred for 1 h at 60 °C. The mixture was cooled down to room temperature, then MeOH was added slowly with ice-water cooling. A white precipitation appeared, and the resulting suspension was stirred and then filtered. The filtered product was washed with water-MeOH. The product was dried under vacuum to give 24.9 g of compound of formula VII (75.2 mmol, yield: 76%). Purity >99.9area% (HPLC).
[0070] In an embodiment, the preparation of hydrochloride salt of (S)-Pirlindole, compound of formula III, was performed as follow.
[0071] In an embodiment, the free amine VII ((S)-8-methyl-3-((S)-l-phenylethyl)-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole) (8,32 g, 25 mmol) was dissolved in DCM (42 mL) and excess of HCI in MeOH (42 mL) was added. The solvents were evaporated under reduced pressure to dryness to give a yellow oil. The residue was dissolved in MeOH (120 mL) and was added to the dispersion of Pd/C (1,74 g, -50% water) in MeOH (20 mL). The reaction mixture was stirred at 50 °C under a 750 KPa (7.5 bar) pressure of hydrogen for 5h. After completion (HPLC) the suspension was filtered through a celite pad, and the filter cake was washed with MeOH. The pH of the resulting solution was checked (<3) and it was evaporated to give the crude hydrochloride salt of compound of formula III. To the crude material iPrOH was added and the suspension was allowed to stir at reflux. The suspensions were filtered, and the product was dried under vacuum to give the hydrochloride salt of (S)-Pirlindole, compound of formula III (5.11 g, 19.5 mmol, yield: 77%). Purity > 99.5% (HPLC). Enantiomeric purity 99.5% (Chiral HPLC). MS (ESI): m/z 227.2 (M+H)+.
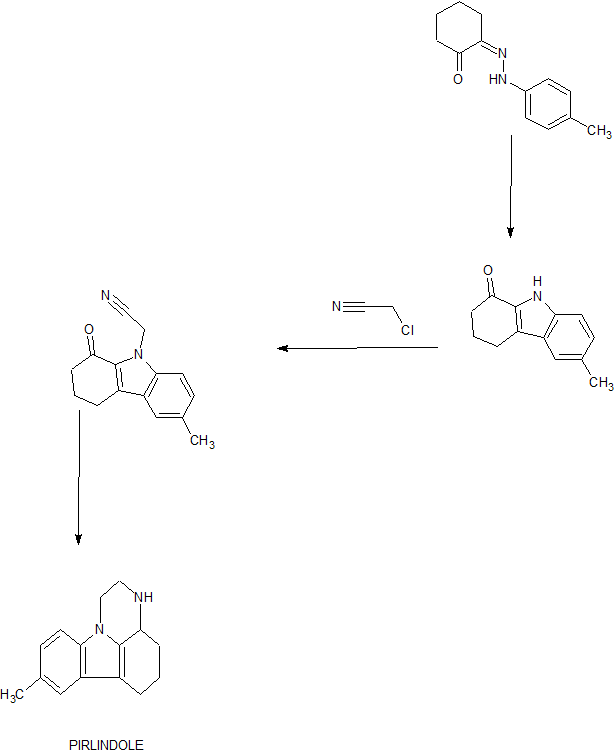
PATENT
Process for the preparation of piperazine ring for the synthesis of pyrazinocarbazole derivatives, such as the antidepressant pirlindole .
Pirlindole hydrochloride is the compound represented in formula I
[0003] It is the common name of 8-methyl-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole hydrochloride which is an active pharmaceutical ingredient marketed with the name Pyrazidol™. The compound is effective as an anti-depressant agent.
[0004] Pirlindole chemical structure belongs to the pyrazinocarbazole group. It is composed of one stereogenic centre which anticipate the existence of two enantiomers, the ( ?)-Pirlindole of formula II and the (S)-Pirlindole of formula III.
[0005] Although Pirlindole pharmacological data and the clinical use were performed on the racemate, recently there have been increasing interest in the pharmacological profile of each enantiomer (WO 2015/171005 Al).
[0006] The document WO 2015/171003Al(Tecnimede group) filed 9th May 2014 discloses a resolution of racemic pirlindole into optically active pirlindole. The Resolution-Racemization-Recycle (RRR) synthesis described involves derivatization by preparation of pairs of diastereomers in the form of salts from an optically active organic acid. These diastereomers can be separated by conventional techniques such as crystallisation. Although it is a very efficient procedure to prepare laboratorial scale or pre-clinical batch of (/?)- or (S)-Pirlindole, it is not economically convenient at an industrial scale because the process relies on Pirlindole racemate as the starting material.
[0007] Processes to prepare Pirlindole involve the formation of a piperazine ring. The state of the art discloses different processes for piperazine ring formation but they are generally a multistep approach, and they are hampered by low yields, expensive reagents, or are reported as unsuccessful (Roderick et al. Journal of Medicinal Chemistry 1966, 9, 181-185).
[0008] The first asymmetric synthesis of Pirlindole enantiomers described by Andreeva et al. (Pharmaceutical Chemistry 1992, 26, 365-369) discloses a one-step process to prepare pyrazinocarbazole piperazine ring system from a tetrahydrocarbazole-amine. The process discloses a very low yield (23.8 %) and employs the use of sodium hydride (NaH) in the presence of dimethyl sulfoxide (DMSO) or dimethyl formamide (DMF), both conditions described as generating exothermic decomposition that can cause reaction ignition or reaction thermal runaway.
[0009] The mixture of sodium hydride with DMSO generates dimsyl anion. This anion is very often used in laboratory scale, but because it is unstable its use on large scale should be under specific precautions. The dimsyl anion decomposition is exothermic. It is reported that dimsyl anion decomposition starts even at 20 °C, and above 40 °C it decomposes at an appreciable rate (Lyness et al. US 3288860).
[0010] The mixture of DMF and sodium hydride is reported in Sax & Lewis’s Dangerous Properties of Industrial Materials to give a violent reaction with ignition above 50 °C. Buckey et al., (Chemical & Engineering News, 1982, 60(28), 5) describes the thermal runaway of a pilot plant reactor containing sodium hydride and DMF from 50 °C. Accelerated Rate Calorimetry (ARC) tests showed exothermic activity as low as 26 °C.
Similar behaviour was also seen with DMA. De Wall et al. (Chem. Eng. News, 1982, 60(37), 5) reports a similar incident, wherein runaway started at 40 °C, and rose 100 °C in less than 10 minutes, boiling off most of the DMF.
[0011] An alternative process for the preparation of a piperazine ring system of a pyrazinocarbazole derivative can involve the formation of a lactam ring in a three steps approach:
1. N-acylation reaction;
2. intramolecular indole acetamide cyclisation to afford a lactam ring;
3. lactam reduction.
[0012] Intramolecular indole chloroacetamide cyclization to yield a lactam ring has been described by Bokanov et al. (Pharmaceutical Chemistry Journal 1988, 23, 12, 1311-1315) particularly in the non-enantioselective synthesis of pyrazinocarbazolone derivatives. Bokanov et al. did not describe the lactam reduction into a piperazine ring.
[0013] Intramolecular indole chloroacetamide cyclization to yield a lactam ring has also been described both by Rubiralta et al. (Journal of Organic Chemistry 54, 23, 5591-5597) and Bennasar, et al. (Journal of Organic Chemistry 1996., 61, 4, 1239-1251), as an unexpected outcome of a photocyclization reaction. The lactam conversion was low (<11% yield).
[0014] Lactam reduction of a pyrazinone into piperazine ring systems is disclosed both by Aubry et al. (Biorganic Medicinal Chemistry Letters 2007, 17, 2598-2602) and Saito et al. (Tetrahedron 1995, 51, 30, 8213-8230) in the total synthesis of alkaloid natural products.
[0015] There exists the need for improved processes for the preparation of piperazine ring derivatives in particular enantioselective processes for the preparation of pyrazinocarbazole intermediates precursors of Pirlindole enantiomers compounds of formula II and III.
Example 1 – Preparation of (S)-8-methyl-3-((S)-l-phenylethyl)-3a,4,5,6-tetrahydro-lH-pyrazino[3,2,l-jk]carbazol-2(3H)-one – Formula IV
[00106] In an embodiment, the preparation of (S)-8-methyl-3-((S)-l-phenylethyl)-3a,4,5,6-tetrahydro-lH-pyrazino[3,2,l-jk]carbazol-2(3H)-one (Formula IV) was carried out as follows. To the solution of VI (S)-6-methyl-N-((S)-l-phenylethyl)-2,3,4,9-tetrahydro-lH-carbazol-l-amine (30 g, 98.5 mmol) in toluene (300 mL), 50 % (w/v) aqueous NaOH (79 g) was added dropwise at 0-5 °C, then the solution of chloroacetyl
chloride (12 mL, 148 mmol, 1.5 equiv.) in toluene (15 mL) was added dropwise at 0-5 °C. The mixture was stirred at 0-5 °C for approximately 2.5 h, and additional chloroacetyl chloride (12 mL, 148 mmol, 1.5 equiv.) in toluene (15 mL) was added dropwise at 0-5 °C. The mixture was stirred at 0-5 °C for approximately 1.5 h. Water was added to the reaction mixture keeping the temperature below 5 °C. The phases were separated, and the aqueous phase was extracted with toluene. The organic phase was treated with 2M aqueous HCI. The resulting suspension was filtered. The filtered solid was identified as the HCI salt of VI, which can be liberated and driven back to the chloroacetylation step. The phases of the mother liquor were separated, and the aqueous phase was extracted with toluene. The organic phase was dried over Na2S04, filtered and concentrated under reduced pressure to about 350 mL as a solution in toluene. The toluene solution of the crude product compound of formula X was reacted in the next step.
[00107] In an embodiment, in the same reaction vessel to the toluene solution of crude intermediate obtained in previous step were added TBAB (0.394 g, 1.22 mmol, 1 w/w% for the theoretical yield of prev. step) and 50 % (w/v) aqueous NaOH (8.1 g, 10 equiv.). The reaction mixture was stirred for 1 h at 65 °C, while the reaction was complete. Water was added to the mixture at 0 °C, and the phases were separated, the organic phase was washed with aqueous HCI, and with water, then dried over Na2S04, filtered and evaporated to give 32.87 g of compound IV (S)-8-methyl-3-((S)-l-phenylethyl)-3a,4,5,6-tetrahydro-lH-pyrazino[3,2,l-jk]carbazol-2(3H)-one (yield: 97% for the two steps) as a brown solid. The crude product was reacted in the next step without further purification.
Example 2 – Preparation of (S)-8-methyl-3-((S)-l-phenylethyl)-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole _ Formula V
[00108] In an embodiment, the preparation of (S)-8-methyl-3-((S)-l-phenylethyl)-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole (Formula V) was performed as follows. To the stirred solution of 32.87 g of IV, (S)-8-methyl-3-((S)-l-phenylethyl)-3a,4,5,6-tetrahydro-lH-pyrazino[3,2,l-jk]carbazol-2(3H)-one (95.4 mmol) in dry THF (170 mL) 66 mL solution of sodium bis(2-methoxyethoxy)aluminium hydride in toluene (70 w/w%, 237 mmol, 2.5 equiv.) was added dropwise. The reaction mixture was warmed to 40 °C, and the end of the addition the mixture was stirred at 50 °C until the total consumption of the starting material. Additional 22 mL of sodium bis(2-methoxyethoxy)aluminium hydride solution (70 w/w%, 79 mmol, 0.8 equiv.) was added dropwise. After completion the mixture was cooled to room temperature and 5% aqueous NaOH was added carefully. Water and DCM were added to the mixture, the phases were separated, and the aqueous phase was extracted with DCM. The organic phase was dried over Na2S04, filtered and the solvent was evaporated to get a brown solid (28.8 g). This crude product was dissolved in DCM and MeOH was added. White solid precipitated. The solid was filtered and washed with MeOH to give V (S)-8-methyl-3-((S)-l-phenylethyl)-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-j‘k]carbazole 14.6 g (yield: 46%) as an off-white cotton-like solid.
Example 3 – Preparation of (S)-Pirlindole Hydrochloride – Formula III
[00109] In an embodiment, the preparation of (S)-Pirlindole hydrochloride III was carried out as follows. The free amine V ((S)-8-methyl-3-((S)-l-phenylethyl)-2,3,3a, 4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole) (8.32 g, 25 mmol) was dissolved in DCM (42 mL) and excess of HCI in MeOH (42 mL) was added. The solvents were evaporated under reduced pressure to dryness to give a yellow oil. The residue was dissolved in MeOH (120 mL) and was added to the dispersion of Pd/C (1.74 g, -50% water) in MeOH (20 mL). The reaction mixture was stirred at 50 °C under 750 KPa (7.5 bar) pressure of hydrogen for 5h. After completion (HPLC) the suspension was filtered through a celite pad, and the filter cake was washed with MeOH. The pH of the resulting solution was checked (<3) and it was evaporated to give the crude hydrochloride salt of compound of formula III. To the crude material iPrOH was added and the suspension was allowed to stir at reflux. The suspensions were filtered, and the product was dried under vacuum to give the hydrochloride salt of (S)-Pirlindole, compound of formula III (5.11 g, 19.5 mmol, yield: 77%). Purity > 99.5% (HPLC). Enantiomeric purity 99.5% (Chiral HPLC). MS (ESI): m/z 227.2 (M+H)+.
[00110] Table 1. Comparative yields
Synthesis Reference
http://www.biomedsearch.com/nih/Pirlindole-in-treatment-depression-meta/21053988.html
General References
- Branco JC, Tome AM, Cruz MR, Filipe A: Pirlindole in the treatment of depression and fibromyalgia syndrome. Clin Drug Investig. 2011 Oct 1;31(10):675-89. doi: 10.2165/11595410-000000000-00000. [PubMed:21877764]
- Bruhwyler J, Liegeois JF, Geczy J: Pirlindole: a selective reversible inhibitor of monoamine oxidase A. A review of its preclinical properties. Pharmacol Res. 1997 Jul;36(1):23-33. doi: 10.1006/phrs.1997.0196. [PubMed:9368911]
- Psychiatry: The State of the Art Volume 3 Pharmacopsychiatry [Link]
- Chemistry Dashboard- Pirlindole [Link]
- Pirlindole in the Treatment of Depression and Fibromyalgia Syndrome [Link]
- Hypertensive effect and cheese [Link]
- Monamine oxide inhibitors [Link]
References
- Jump up^ Medvedev AE, et al. The influence of the antidepressant pirlindole and its dehydro-derivative on the activity of monoamine oxidase A and GABAA receptor binding. Chapter 36 in MAO – The Mother of all Amine Oxidases (Journal of Neural Transmission. Supplementa). Eds Finberg JPM, Youdim MBH, Riederer P, Tipton KF. Special edition of Journal of Neural Transmission, Suppl. 52 1st ed. 1998 ISBN 978-3211830376
 |
|
| Clinical data | |
|---|---|
| Trade names | Pirazidol |
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 20–30% |
| Protein binding | 95% |
| Metabolism | hepatic |
| Onset of action | 2 to 8 hours |
| Elimination half-life | 185 hours |
| Excretion | urine (50–70%), feces (25–45%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C15H18N2 |
| Molar mass | 226.32 g/mol |
| 3D model (JSmol) | |
//////////////Pirlindole, DEPRESSION, Pyrazidol, 60762-57-4
CC1=CC2=C(C=C1)N3CCNC4C3=C2CCC4