














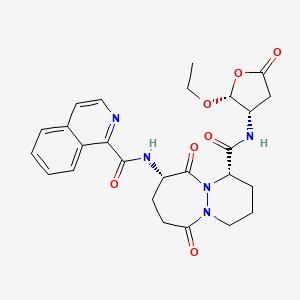
Pralnacasan
VX 740
cas 192755-52-5
(4S,7S)-N-[(2R,3S)-2-ethoxy-5-oxooxolan-3-yl]-7-(isoquinoline-1-carbonylamino)-6,10-dioxo-2,3,4,7,8,9-hexahydro-1H-pyridazino[1,2-a]diazepine-4-carboxamide
N-[(4S,7S)-4-{[(2R,3S)-2-ethoxy-5-oxooxolan-3-yl]carbamoyl}-6,10-dioxo-octahydro-1H-pyridazino[1,2-a][1,2]diazepin-7-yl]isoquinoline-1-carboxamide
(1S,9S)-N-((2R,3S)-2-Ethoxy-5-oxotetrahydrofuran-3-yl)-9-((isoquinolin-1-ylcarbonyl)amino)-6,10-dioxooctahydro-6-H-pyridazino(1,2-a)(1,2)diazepine-1-carboxamide
6H-Pyridazino(1,2-a)(1,2)diazepine-1-carboxamide, N-((2R,3S)-2-ethoxytetrahydro-5-oxo-3-furanyl)octahydro-9-((1-isoquinolinylcarbonyl)amino)-6,10-dioxo-, (1S,9S)-
- HMR 3480
- HMR3480
- HMR3480/VX-740
- Pralnacasan
- UNII-N986NI319S
- VX 470
- VX-740
C26H29N5O7, 523.543
NSAID, ICE inhibitor & metastasis inhibitor.

Abstract
Novel 1-(2-acylhydrazinocarbonyl)cycloalkyl carboxamides were designed as peptidomimetic inhibitors of interleukin-1β converting enzyme (ICE). A short synthesis was developed and moderately potent ICE inhibitors were identified (IC50 values <100 nM). Most of the synthesized examples were selective for ICE versus the related cysteine proteases caspase-3 and caspase-8, although several dual-acting inhibitors of ICE and caspase-8 were identified. Several of the more potent ICE inhibitors were also shown to inhibit IL-1β production in a whole cell assay (IC50 < 500 nM).
Graphical abstract
Novel 1-(2-acylhydrazinocarbonyl)cycloalkyl carboxamides were designed and synthesized as selective peptidomimetic inhibitors of interleukin-1β converting enzyme (ICE IC50 values <100 nM).

Abstract

Peptides containing N2-acyl piperazic or 1,6-dehydropiperazic acids can be formed efficiently via a novel multicomponent reaction of 1,4,5,6-tetrahydropyridazines, isocyanides, and carboxylic acids. Remarkably, the reaction’s induced intramolecularity can enable the regiospecific formation of products with N2-acyl piperazic acid, which counters the intrinsic and troublesome propensity for piperazic acids to react at N1 in acylations. The utility of the methodology is demonstrated in the synthesis of the bicyclic core of the interleukin-1β converting enzyme inhibitor, Pralnacasan.





substituent is




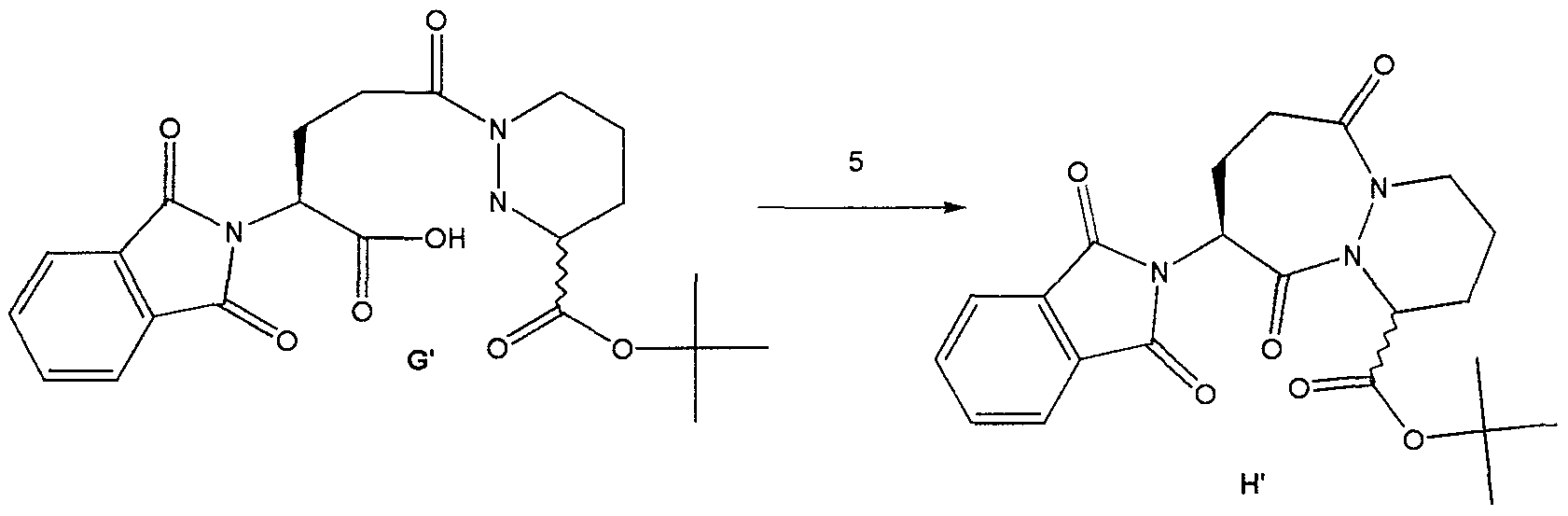


t-Butyl-9- (isoquinolin-1-oylamino) -6, 10-dioxo- 1,2,3,4,7,8,9, 10-octahydro-6-H-pyridazino [ 1 , 2-a] [1,2] diazepine-1-carboxylate To a solution of the amine I’ (79 g, 0.265 mol) and isoquinolin-1-carboxylic acid (56g, 0.32 mol) in dichloromethane : DMF (400mL: 400mL) was added hydroxybenztriazole (54 g, 0.4 mol) and l-(3- dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (74 g, 0.39 mol) and the resulting mixture was allowed to stir at ambient temperature for 16 hours. The reaction mixture was poured into water and extracted with ethyl acetate. The ethyl acetate layer was washed with 0.5N sodium bisulfate, water, sodium bicarbonate, brine, dried over sodium sulfate and concentrated in vacuo to afford 122 g of compound J’ as an orange solid-foam.








Hθ

The concentrate (B) was taken up in n-butanol
Example 5 Conversion of Intermediate IV to Intermediate Vl-a Cbzy

A. Phthalimide removal to form IX-b




[1S, 9S (2RS,3S) ]N- (2-benzyloxy-5-oxotetrahydrofuran-3- yl) -6,10-dioxo-9- (isoquinolin-1-oylamino) – 1,2,3,4,7,8,9, 10-octahydro-6-H-pyridazino [1 , 2-a] [1,2] diazepine-1-carboxamide (4-b) : To a solution of (3S, 2RS) 3-allyloxycarbonylamino-2-benzyloxy-5-oxotetrahydrofuran [Bioorq. & Med. Chem. Lett., 2, pp. 615-618 (1992)] (4.4 g, 15.1 mmol) in dichloromethane was added N,N- dimethylbarbituric acid (5.9g, 3.8 mmol) then tetrakispalladium(O) triphenyl phosphine (1.7 g, 1.5 mmol) and the resulting mixture was allowed to stir at ambient temperature for 15 minutes. To the resulting mixture was added the acid, compound XIII (from step C) (5.0 g, 12.6 mmol), hydroxybenzotriazole (2.0 g, 14.8 mmol), then 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (2.7g, 14 mmol) and the reaction was allowed to stir for 3 hours at ambient temperature. The reaction mixture was then poured into water and extracted with ethyl acetate. The organics were washed with 0.5M sodium bisulfate, water, sodium bicarbonate, brine, dried over magnesium sulfate and concentrated in vacuo to afford 2.6 g of the crude product as a yellow foam. The crude material was purified by column chromatography (Si02, dichloromethane: acetone 9:1 – 3:1) to afford 1.2 g of the compound 4-b. Compounds of formulae VII and VIII, and related compounds, that may be synthesized using the method of this invention as an intermediate step are described in WO 97/22619 and United States Patent 6,204,261 the disclosure of which is herein incorporated by reference. Those related compounds may be synthesized from the product of the method of this invention, I, IV, or V, through modifications of the procedure set forth in Examples 4 through 6. Such modifications are well known in the art.
Patent
////////////////Pralnacasan, VX 740, VX 470, HMR 3480, пралнаказан , برالناكاسان , 普那卡生 ,
CCOC1C(CC(=O)O1)NC(=O)C2CCCN3N2C(=O)C(CCC3=O)NC(=O)C4=NC=CC5=CC=CC=C54