















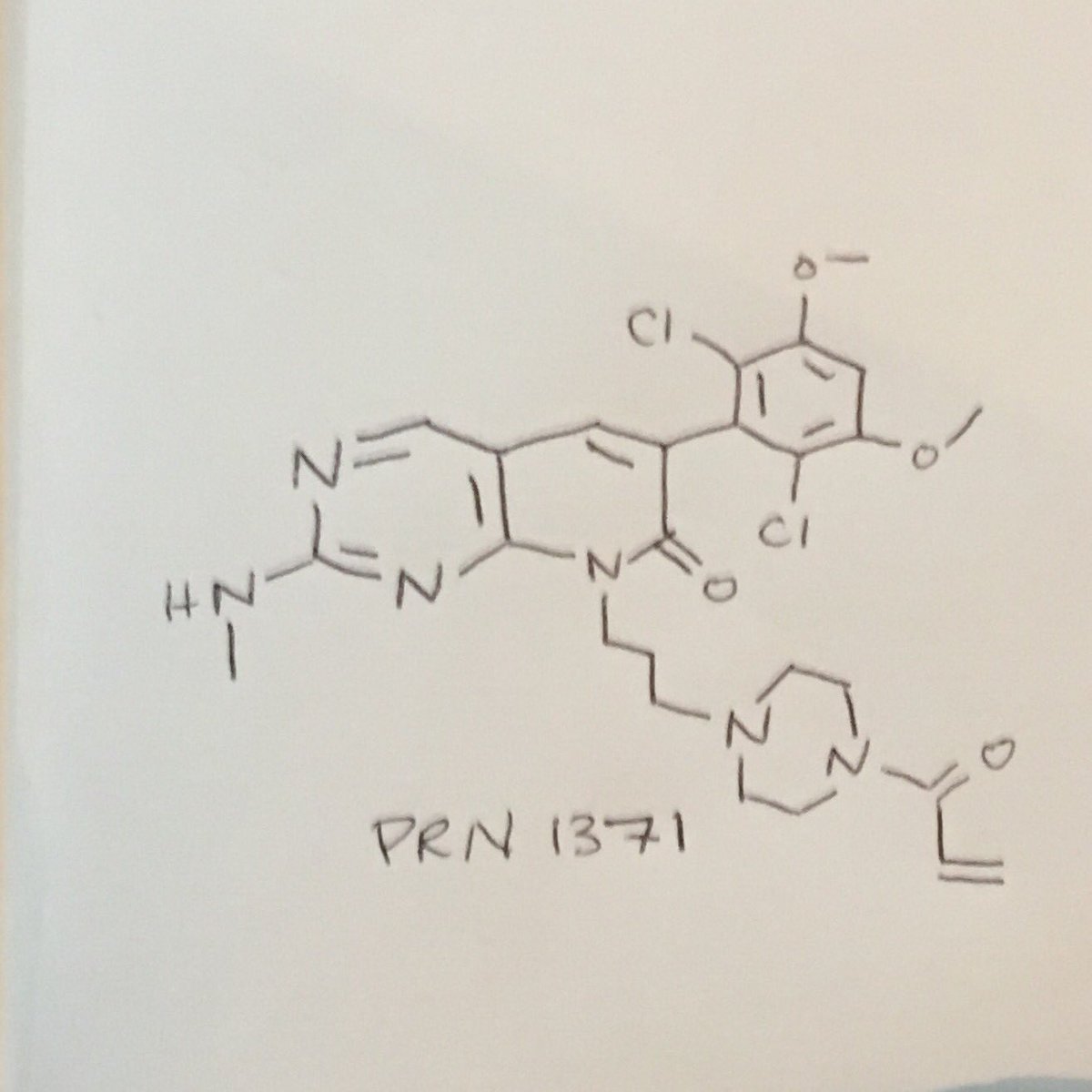
PRN 1371
- Molecular Formula C26H30Cl2N6O4
- Average mass 561.460
cas 1802929-43-6
8-[3-(4-Acryloyl-1-
6-(2,6-Dichloro-3,5-dimethoxyphenyl)-2-(methylamino)-8-[3-[4-(1-oxo-2-propen-1-yl)-1-piperazinyl]propyl]pyrido[2,3-d]pyrimidin-7(8H)-one
Phase I Solid tumours
- Originator Principia Biopharma
- Class Small molecules
- Mechanism of Action Fibroblast growth factor receptor antagonists
- 06 Jun 2016 Adverse events data from a phase I trial in Solid tumours presented at the 52nd Annual Meeting of the American Society of Clinical Oncology (ASCO- 2016)
- 01 Nov 2015 Phase-I clinical trials in Solid tumours in USA (PO) (NCT02608125)
- 12 Jan 2015 Preclinical trials in Cancer in USA (PO)
| Inventors | Erik Verner, Kenneth Albert Brameld |
| Applicant | Principia Biopharma, Inc. |


Erik Verner


CONTD………………..

Fibroblast growth factors (FGFs) and their receptors (FGFRs) play important roles in physiological processes relating to tissue repair, hematopoiesis, bone growth, angiogenesis and other aspects of embryonic development. Alterations in the FGF signaling pathway have also emerged as important drivers in human disease. FGF signaling can be deregulated through multiple mechanisms, including gene amplification, activating mutations and translocations, overexpression, altered FGFR gene splicing, and autocrine or paracrine overproduction of the ligands of FGFR. Deregulated FGF signaling has been documented in human tumors, including breast (see Ray, M. E., et. al., 2004. Genomic and expression analysis of the 8pl 1-12 amplicon in human breast cancer cell lines. Cancer Res 64:40-47), multiple myeloma (see Keats, J.J., et. al., 2006. Ten years and counting: so what do we know about t(4;14)(pl6;q32) multiple myeloma. Leuk Lymphoma 47:2289-2300), non-invasive bladder (see Billerey, C, et al. 2001. Frequent
FGFR3 mutations in papillary non-invasive bladder (pTa) tumors. Am J Pathol 158: 1955-1959), endometrial (see Pollock, P.M., et al. 2007. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene 26:7158-7162), gastric (see Jang, J.H., et. al, 2001. Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers. Cancer Res 61 :3541-3543), prostate cancers (see Sahadevan, K., D et. al., 2007. Selective over-expression of fibroblast growth factor receptors 1 and 4 in clinical prostate cancer. J Pathol 213:82-90), lung (see Hammerman P, et al. Genomic characterization and targeted therapeutics in squamous cell lung cancer [abstract]; Proceedings of the 14th World Conference on Lung Cancer; 2011 3-7 July; Aurora (CO); and International Association for the Study of Lung Cancer; 2011), esophageal (see Hanada K, et al, Identification of fibroblast growth factor-5 as an overexpressed anti-gen in multiple human adenocarcinomas. Cancer Res 2001; 61 : 5511-6), cholangiocarcinoma (see Arai, Y., et al. 2014. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 59, 1427-1434 and Borad, M. J., et al. 2014). Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS genetics 10, el004135), glioblastoma (see Rand V., et. al. Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc Natl Acad Sci U S A 2005; 102: 14344 – 9 and Parker, et. al. 2014. Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours. The Journal of pathology 232, 4-15). FGFR1 translocations and FGFR1 fusions are frequently observed in 8pl 1 myeloproliferative syndromes (Jackson, C. C, Medeiros, L. J., and Miranda, R. N. (2010). 8pl 1 myeloproliferative syndrome: a review. Human pathology 41, 461-476). Activating mutations in FGFR3 have been shown to cause a number of dwarf syndromes (see Harada, D., et. al, 2009. FGFR3-related dwarfism and cell signaling. J Bone Miner Metab 27:9-15) including achondroplasia (see Bellus, G.A., et. al., 1995. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet 56:368-373; Bellus, G.A., et. al., 1995. A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nat Genet 10:357-359; and Rousseau, F., et. al, 1994. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature 371 :252-254), Crouzon dermoskeletal syndromes (see Robin, N.H., et. al, 1993. FGFR-Related Craniosynostosis Syndromes), hyopochondroplasia (see Prinos, P., et. al., 1995. A common FGFR3 gene mutation in hypochondroplasia. Hum Mol Genet 4:2097-2101), Muenke syndrome (see Muenke, M., et al. 1997. A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet 60:555-564), SADDAN (severe achondroplasia with developmental delay and acanthosis nigricans) (see Bellus, G.A., et al. 1999. Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN): phenotypic analysis of a new skeletal dysplasia caused by a Lys650Met mutation in fibroblast growth factor receptor 3. Am J Med Genet 85:53-65;
Tavormina, P.L., et al. 1999. A novel skeletal dysplasia with developmental delay and acanthosis nigricans is caused by a Lys650Met mutation in the fibroblast growth factor receptor 3 gene. Am J Hum Genet 64:722-731), thanatophoric dysplasia ( see dAvis, P.Y., et. al, 1998. Constitutive activation of fibroblast growth factor receptor 3 by mutations responsible for the lethal skeletal dysplasia thanatophoric dysplasia type I. Cell Growth Differ 9:71-78; Kitoh, H., et. al, 1998. Lys650Met substitution in the tyrosine kinase domain of the fibroblast growth factor receptor gene causes thanatophoric dysplasia Type I. Mutations in brief no. 199. Online. Hum Mutat 12:362- 363; and Tavormina, P.L., et. al, 1995. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat Genet 9:321-328), platyspondylic lethal skeletal dysplasia (see Brodie, S.G., et. al, 1999. Platyspondylic lethal skeletal dysplasia, San Diego type, is caused by FGFR3 mutations. Am J Med Genet 84:476-480), and cervical cancer (see Cappellen, D., et. al., 1999. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet 23: 18-20). Activating mutations in FGFR4 have been identified in rhabdomyosarcoma (see Shukla, N., et. al, Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin Cancer Res 18:748-757 and Marshall, A.D., et. al, PAX3-FOX01 and FGFR4 in alveolar rhabdomyosarcoma. Mol Carcinog 51 :807-815). For these reasons, FGFRs are attractive therapeutic target for the treatment of diseases.
Patent
WO 2015120049
Example 6
Synthesis of 8-(3-(4-acryloylpiperazin-l-yl)propyl)-6-(2,6-dichloro-3,5-dimethoxyphenyl)-2- (methylamino)pyrido[2,3-d]pyrimidin-7(8H)-one

Step 1
To a solution of 3-(piperazin-l-yl)propan-l-ol (1 g, 6.93 mmol, 1.00 equiv) in THF (50 mL) and TEA (2 g) was added di-tert-butyl dicarbonate (2.26 g, 10.36 mmol, 1.49 equiv). The resulting solution was stirred for 2 h at room temperature and then concentrated. The residue was purified by chromatography (DCM/MeOH (15: 1)) to provide 1.48 g (87%) of tert-butyl 4-(3-hydroxypropyl)piperazine-l-carboxylate as a light yellow liquid.
Step 2
To a solution of tert-butyl 4-(3-hydroxypropyl)piperazine-l-carboxylate (1.48 g, 6.06 mmol, 1.00 equiv) in DCM (60 mL), imidazole (620 mg) and TPP (2.38 g, 9.07 mmol, 1.50 equiv) was added I2 (2.31 g, 9.10 mmol, 1.50 equiv). The resulting solution was stirred for 2 h at room temperature and then concentrated. The residue was purified by chromatography
(DCM/MeOH (50: 1)) to provide 1.65 g (77%) of tert-butyl 4-(3-iodopropyl)piperazine-l-carboxylate as yellow oil.
Step 3
To a solution of 6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylsulfanyl)-7H,8H-pyrido[2,3-d]pyrimidin-7-one (600 mg, 1.51 mmol, 1.00 equiv) in acetone (50 mL) and K2C03 (630 mg) was added tert-butyl 4-(3-iodopropyl)piperazine-l-carboxylate (640 mg, 1.81 mmol, 1.20 equiv). The resulting solution was heated to reflux for 3 h and then the solids were filtered out. The residue was purified by chromatography (DCM/EtOAc (2:1)) to provide 720 mg (77%) of tert-butyl 4-[3-[6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylsulfanyl)-7-oxo-7H,8H-pyrido[2,3-d]pyrimidin-8-yl]propyl]piperazine-l-carboxylate as a yellow solid.
Step 4
To a solution of tert-butyl 4-[3-[6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methyl-sulfanyl)-7-oxo-7H,8H-pyrido[2,3-d]pyrimidin-8-yl]propyl]piperazine-l-carboxylate (720 mg, 1.15 mmol, 1.00 equiv) in CHC13 (50 mL) was added mCPBA (600 mg). The resulting solution was stirred overnight at room temperature and then quenched with sat. Na2C03. The resulting solution was extracted DCM/MeOH(10: l) and the organic layer was concentrated. This provided 750 mg (97%)) of 4-[(tert-butoxy)carbonyl]-l-[3-[6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-
methanesulfonyl-7-oxo-7H,8H-pyrido[2,3-d]pyrimidin-8-yl]propyl]piperazin- 1 -ium- 1 -olate as a yellow solid.
Step 5
To a solution of 4-[(tert-butoxy)carbonyl]-l-[3-[6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-methanesulfonyl-7-oxo-7H,8H-pyrido[2,3-d]pyrimidin-8-yl]propyl]piperazin- 1 -ium- 1 -olate (750 mg, 1.12 mmol, 1.00 equiv) in tert-BuOH (50 mL), was added MeNH2/THF(2N) (1 mL). The resulting solution was stirred for 2 h at 60° C and then concentrated. This provided 680 mg (98%) of 4-[(tert-butoxy)carbonyl]-l-[3-[6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)-7-oxo-7H,8H-pyrido[2,3-d]pyrimidin-8-yl]propyl]piperazin-l-ium-l-olate as a yellow solid.
Step 6
To a solution of 4-[(tert-butoxy)carbonyl]-l-[3-[6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)-7-oxo-7H,8H-pyrido[2,3-d]pyrimidin-8-yl]propyl]piperazin-l-ium-l-olate (680 mg, 1.09 mmol, 1.00 equiv) in MeOH (100 mL) was added Zn (1 g) and sat. NH4C1 (4 mL). The resulting reaction mixture was stirred overnight at room temperature and then solids were filtered out. The residue was purified by chromatography (DCM/MeOH (35: 1)) to provide 650 mg (98%) of tert-butyl 4-[3-[6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)-7-oxo-7H,8H-pyrido[2,3-d]pyrimidin-8-yl]propyl]piperazine-l-carboxylate as a yellow solid.
Step 7
To a solution of tert-butyl 4-[3-[6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)-7-oxo-7H,8H-pyrido[2,3-d]pyrimidin-8-yl]propyl]piperazine-l-carboxylate (650 mg, 1.07 mmol, 1.00 equiv) in dioxane (12 mL), was added cone. HC1 (3 mL). The resulting solution was stirred for 3 h at room temperature and then concentrated. This provided 550 mg (95%) of 6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)-8-(3-(piperazin-l-yl)propyl)pyrido[2,3-d]pyrimidin-7(8H)-one hydrochloride as an off-white solid.
Step 8
To a solution of 6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)-8-[3-(piperazin-l-yl)propyl]-7H,8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride (250 mg, 0.49 mmol, 1.00 equiv) in DCM (20 mL) was added TEA (120 mg, 1.19 mmol, 2.41 equiv) and prop-2-enoyl chloride (54 mg, 0.60 mmol, 1.21 equiv). The resulting solution was stirred for 2 h at room temperature and then quenched with H20 (30 mL). The resulting solution was extracted with DCM/MeOH(10:l) and the organic layers combined and concentrated. The crude product was purified by Prep-HPLC (Column, SunFire Prep CI 8 OBD Column, 150mm 5um lOnm; mobile phase, Water with lOmmol NH4HC03and MeCN (30.0% MeCN up to 80.0% in 10 min);
Detector, nm). This provided 112.1 mg (41%>) of 6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-
(methylamino)-8-[3-[4-(prop-2-enoyl)piperazm^
one as a white solid. MS (ESI, pos. ion) m/z: 561.1 (M+l).
PATENT
Example 1
Synthesis of Compound (I)
Step 1
2-(3,5-Dimethoxyphenyl)acetic acid (1000 g) was charged into appropriately sized three-neck RBF equipped with a condenser and dissolved with methanol (10 L). Concentrated sulfuric acid (20 g) was added and a solution was brought to gentle boiling. Reaction progress was monitored by HPLC. The reaction mixture was transferred to appropriately sized RBF and
concentrated to ca. 3 L. and then co-evaporated with DMSO (3 L) to about 4 L and the residue containing methyl 2-(3,5-dimethoxyphenyl)acetate (1071 g) was telescoped to Step 2.
Step 2
To an appropriate reactor equipped with mechanical stirrer methyl 2-(3,5-dimethoxyphenyl)acetate (1071 g) in DMSO (3.2 L), 4-amino-2-(methylthio)-pyrimidine-5-carbaldehyde (819 g, 0.95 eq.), potassium carbonate (1057 g, 1.5 eq.) and cesium carbonate (249 g, 0.15 eq.) was charged and the mixture was stirred at 50 °C. After 15 h, the mixture containing 6-(3,5-dimethoxyphenyl)-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one was cooled to RT. Potassium carbonate (854g, 1.2 eq.) and tert-butyl 4-(3 -((methyl sulfonyl)oxy )propyl)piperazine-1-carboxylate HC1 (2112 g, 1.1 eq.) was charged. Upon completion of ther eaction, ethyl acetate and water were added.
Organic layer was separated and aqueous layer was extracted with ethyl acetate.
Combined organic layers were washed with 25% aqueous solution of sodium chloride. Organic phase was dried over anhydrous magnesium sulfate. Drying agent was filtered off and washed with ethyl acetate. The filtrate was concentrated to ca. 9.6 L. and cooled to 0-5°C. A solution of ^-toluenesulfonic acid (970 g, 1.0 eq.) in ethyl acetate (4.28 L) was added dropwise. The resulted suspension was slowly warmed to RT and stirred for 5 h. Solids were filtered off, washed with ethyl acetate and dried give tert-butyl-4-(3-(6-(3,5-dimethoxyphenyl)-2-(methylthio)-7-oxopyrido[2,3-d]pyrimidin-8(7H)-yl)propyl)piperazine- 1-carboxylate 4-methylbenzenesulfonate. Step 3
To an appropriate reactor equipped with mechanical stirrer was charged acetic acid (12 L), 6-(3,5-dimethoxyphenyl)-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (2000 g) and triethylamine (639 g, 2.3 eq.). Internal temperature was adjusted to approximately 20°C and N-chlorosuccinimide (1651 g, 4.5 eq.) was added at 20-30°C. Reaction was stirred for 2 hours. Ethyl acetate (30 L) was added. 5% aqueous NaCl solution (20 L) was added. The organic layer was separated and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with 30 % aqueous potassium carbonate solution (14 L). The organic layer was concentrated to ~ 12 L and used for next step directly.
Step 4
To tert-butyl-4-(3-(6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylsulfonyl)-7-oxopyrido[2,3-d]pyrimidin-8(7H)-yl)propyl)piperazine- 1-carboxylate (1804 g) in ethyl acetate extract (12 L)from Step 3, was added 2M methylamine solution in THF (3435 mL) was slowly added maintaining temperature below 30°C. After reaction was complete, the suspension concentrated to 3.3 L and ethyl acetate (6 L) was added. The mixture was heated at 50°C for 2h, and then cooled to RT. Solids were filtered off and washed with ethyl acetate, water and dried to give tert-butyl-4-(3-(6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)-7-oxopyrido[2,3-d]pyrimidin-8(7H)-yl)propyl)piperazine-l-carboxylate (1845 g).
Step 5
tert-Butyl-4-(3-(6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)-7-oxo-pyrido[2,3-d]pyrimidin-8(7H)-yl)propyl)piperazine-l-carboxylate (125 g) was charged into appropriately sized three-neck RBF equipped with a condenser and suspended in acetone (1000 mL). Concentrated (36%) aqueous hydrochloric acid (100 mL) was slowly added and the mixture was heated to 45°C for 1 h. the reaction mixture was gradually cooled to RT over 4 h and filtered, washed with acetone and dried to give tert-butyl-4-(3-(6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)-7-oxopyrido[2,3-d]pyrimidin-8(7H)-yl)propyl)piperazine-l-carboxylate»3HCl (125 g) in 98% yield.
Step 6
To an appropriate reactor tert-butyl-4-(3-(6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)-7-oxopyrido[2,3-d]pyrimidin-8(7H)-yl)propyl)piperazine-l-carboxylate (50 g) and DMF (500 mL) was charged while stirring at RT. The suspension was cooled to 0-5°C and saturated aqueous sodium bicarbonate solution (375 mL) was slowly added maintaining temperature below 15°C with emission of C02. The mixture was cooled again to 0-5°C and acryloyl chloride (8.6 mL, 1.3 eq.) was slowly added at temperature below 10°C. Once acryloyl chloride addition was finished the reaction mixture was gradually warmed to RT over 1 h.
Saturated aqueous sodium bicarbonate solution (75 mL) was slowly added and the resulted mixture was heated at 45-55°C for 0.5-1.5 h. It was then gradually cooled to RT and stirred for another 0.5-1.5 h. Solids were filtered off, washed with water and dried.
Crude product was dissolved in dichloromethane (750 mL) at reflux and the solution was cooled to ambient temperature. Silica gel (7.5 g) was added while stirring. After 30 min. the mixture was filtered through Celite and the filtering bed was washed with dichloromethane.
Ethyl acetate (250 mL) was added and the solution was concentrated under reduced to about 250 mL at 40 – 50 °C. Ethyl acetate (450 mL) was slowly added at 50°C. After 30 min. the suspension was slowly cooled to 40°C and solids were filtered off, washed with ethyl acetate and dried to give 36 g of 8-(3-(4-acryloylpiperazin-l-yl)propyl)-6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)pyrido[2,3-d]pyrimidin-7(8H)-one in 82%. XRPD analysis of the product showed an XRPD pattern for a highly crystalline compound, which was assigned as Form 1 (discussed in further detail below).
///////////PRN 1371, Phase I, Solid tumours, Principia Biopharma
Clc1c(OC)cc(OC)c(Cl)c1C4=Cc2cnc(NC)nc2N(CCCN3CCN(CC3)C(=O)C=C)C4=O
Now in #MEDI 1st time disclosures Principia Biopharma’s Kenneth Brameld on another FGFR inhibitor for solid tumors #ACSSanFran