KIN-3248 is a small molecule that targets and inhibits oncogenic fibroblast growth factor receptors (FGFRs). It was designed to mainly target FGFR2 and FGFR3 alterations, which act as oncogenic drivers in 10-20% of cholangiocarcinoma and 20-35% of urothelial cancers, respectively. While effective, disease progression may occur 6 to 8 months after treatment with currently approved FGFR inhibitors is started, and this effect is usually associated with on-target resistance mutations in the kinase domain of FGFR. Therefore, the broad inhibition of FGFR isoforms may be effective against different types of tumors. The safety, tolerability, pharmacokinetics, and preliminary efficacy of KIN-3248 are currently being evaluated in adults with advanced tumors harboring FGFR2 and/or FGFR3 gene alterations. In February 2023, Kinnate Biopharma received Fast Track designation from the FDA for KIN-3248 to treat unresectable, locally advanced or metastatic cholangiocarcinoma (CCA).
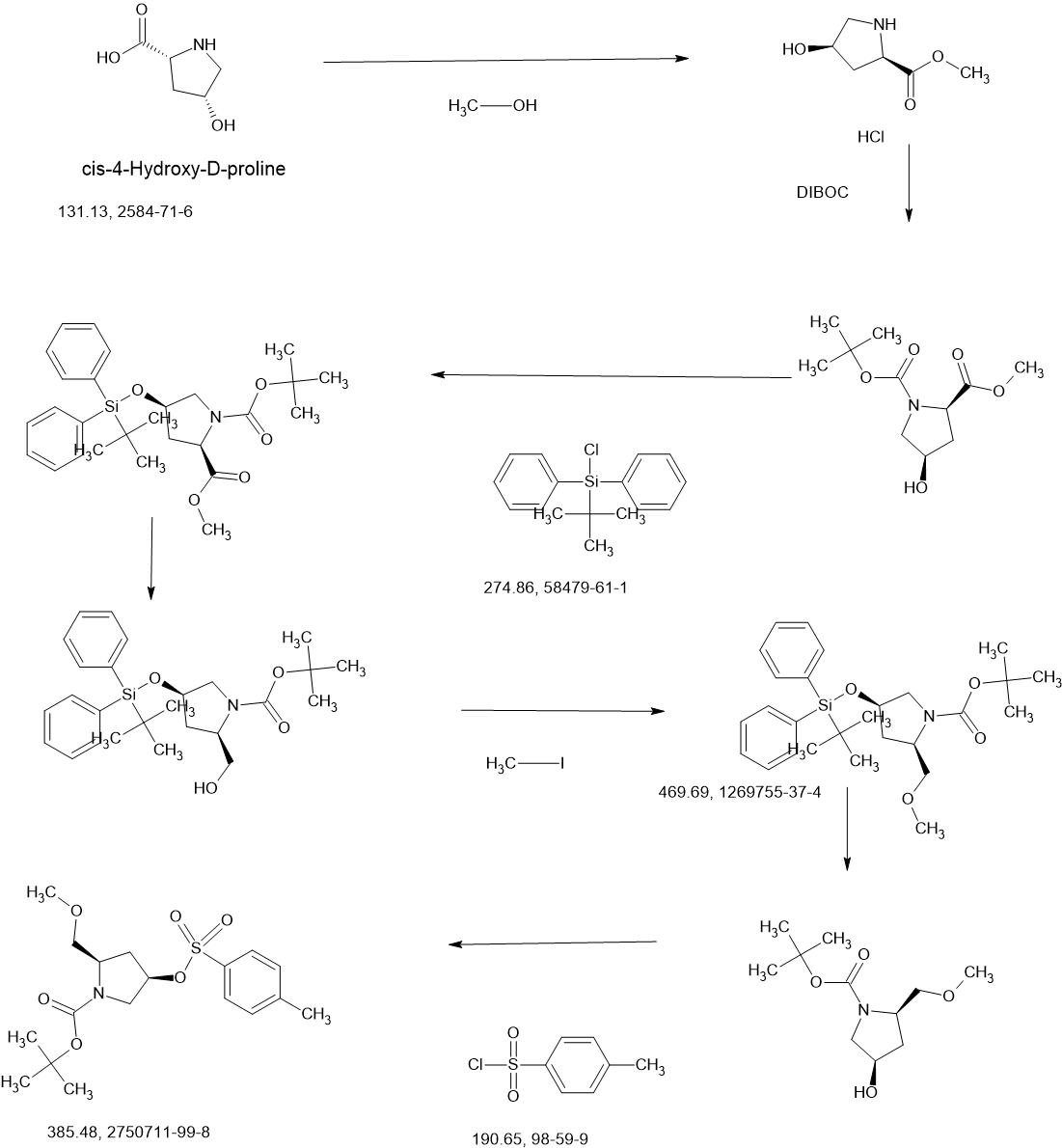
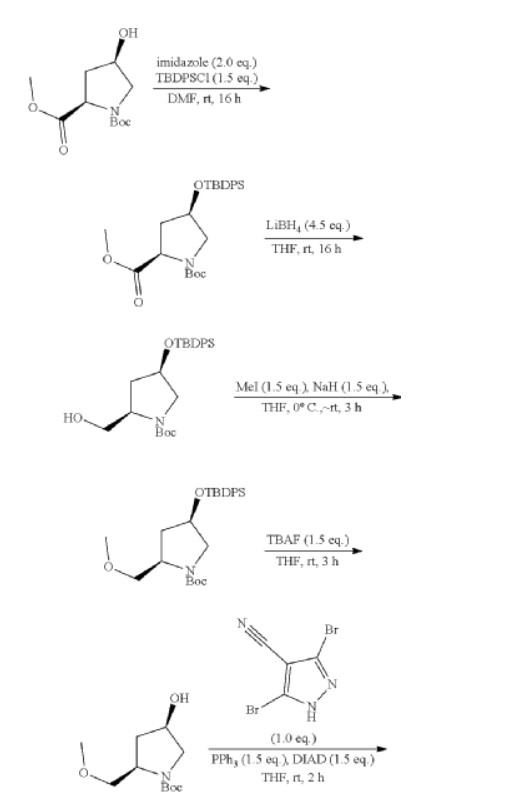
To a stirred solution of 1-(tert-butyl) 2-methyl (2R,4R)-4-hydroxypyrrolidine-1,2-dicarboxylate (8.00 g, 32.62 mmol) and imidazole (4.44 g, 65.23 mmol) in DMF (80.00 mL) was added tert-butyl(chloro)diphenylsilane (13.45 g, 48.93 mmol) at 0° C. over 30 min. The reaction mixture was stirred for 16 h at room temperature. The resulting mixture was diluted with water (400 mL) and extracted with EA (3×300 mL). The combined organic layers was washed with brine (5×500 mL), dried over anhydrous Na 2SO 4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with PE/EA (6/1). The fractions contained desired product were combined and concentrated to afford 1-(tert-butyl) 2-methyl (2R,4R)-4-((tert-butyldiphenylsilyl)oxy)pyrrolidine-1,2-dicarboxylate (14.20 g, 90%) as a colorless oil. MS ESI calculated for C 27H 37NO 5Si [M+H] +, 484.24, found 484.25.
To a stirred solution of 1-(tert-butyl) 2-methyl (2R,4R)-4-((tert-butyldiphenylsilyl)oxy)pyrrolidine-1,2-dicarboxylate (30.00 g, 62.02 mmol) in THF (300.00 mL) was added LiBFi 4 (6.08 g, 0.28 mol) in portions at 0° C. under nitrogen atmosphere. The reaction mixture was stirred for 16 h at room temperature under nitrogen atmosphere. The resulting mixture was acidified to pH 5 with HCl (1M) at 0° C. and then basified to pH 8 with saturated NaHCO 3 (aq.). The resulting mixture was extracted with EA (4×500 mL). The combined organic layers was dried over anhydrous Na 2SO 4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with PE/EA (5/1). The fractions contained desired product were combined and concentrated to afford tert-butyl (2R,4R)-4-[(tert-butyldiphenylsilyl)oxy]-2-(hydroxymethyl)pynolidine-1-carboxylate (24.00 g, 85%) as a light yellow oil. MS ESI calculated for C 26H 37NO 4Si [M+H] +, 456.25, found 456.30.
To a suspension of NaH (0.20 g, 8.33 mmol) in THF (18 mL) was added a solution of tert-butyl (2R,4R)-4-[(tert-butyldiphenylsilyl)oxy]-2-(hydroxymethyl)pyrrolidine-1-carboxylate (2.50 g, 5.49 mmol) in THF (64.00 mL) slowly at 0° C. under nitrogen atmosphere. After stirred at 0° C. for 1 h, to the above mixture was added CH 3I (1.17 g, 8.23 mmol) dropwise at 0° C. The reaction mixture was stirred for additional 3 h at room temperature. The resulting mixture was diluted with water (60 mL), and then extracted with EA (3×30 mL). The combined organic layers was dried over anhydrous Na 2SO 4 and filtered. The filtrate was concentrated under reduced pressure to afford tert-butyl (2R,4R)-4-[(tert-butyldiphenylsilyl)oxyl]-2-(methoxymethyl)pyrrolidine-1-carboxylate (2.30 g, 89%) as a light yellow solid. MS ESI calculated for C 27H 39NO 4Si [M+H] +, 470.26, found 470.30.
To a stirred solution of tert-butyl (2R,4R)-4-[(tert-butyldiphenylsilyl)oxy]-2-(methoxymethyl)pyrrolidine-1-carboxylate (46.30 g, 98.57 mmol) in THF (375.00 mL) was added tetra-n-butylammonium fluoride (1 M in THF) (146.70 mL, 0.14 mol) at 0° C. The reaction mixture was stirred at room temperature for 3 h. The resulting mixture was diluted with water (1 L) and extracted with EA (3×500 mL). The combined organic layers was dried over anhydrous Na 2SO 4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with PE/EA (3/1). The fractions contained desired product were combined and concentrated to afford tert-butyl (2R,4R)-4-hydroxy-2-(methoxymethyl)pyrrolidine-1-carboxylate (16.60 g, 73%) as a light yellow oil. MS ESI calculated for C 11H 21NO 4 [M+H] +, 232.15, found 232.20.
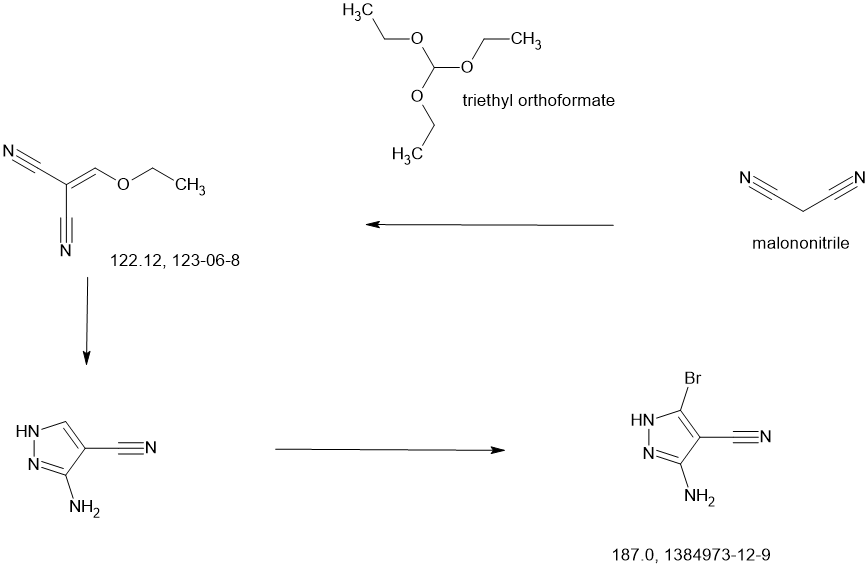
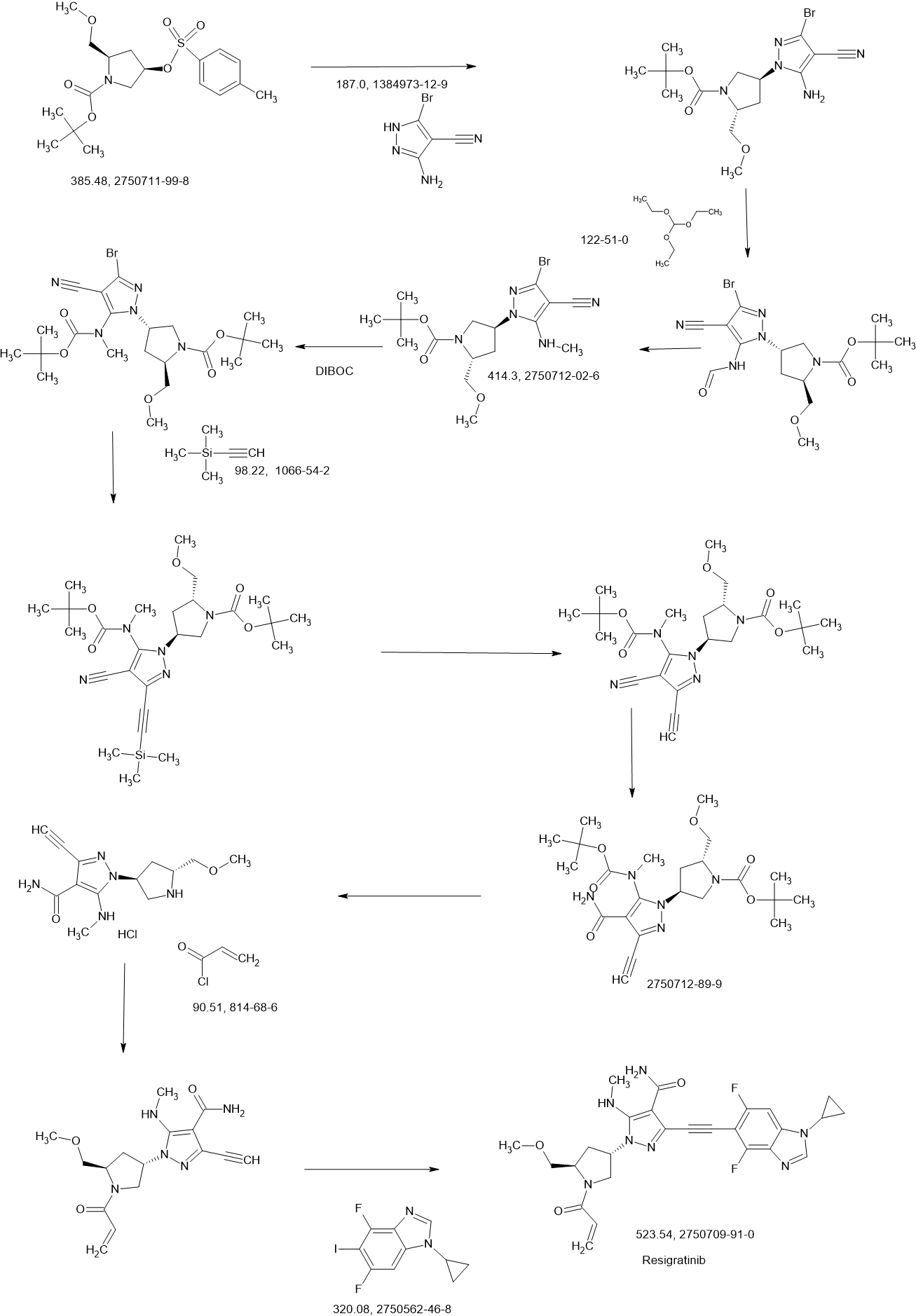
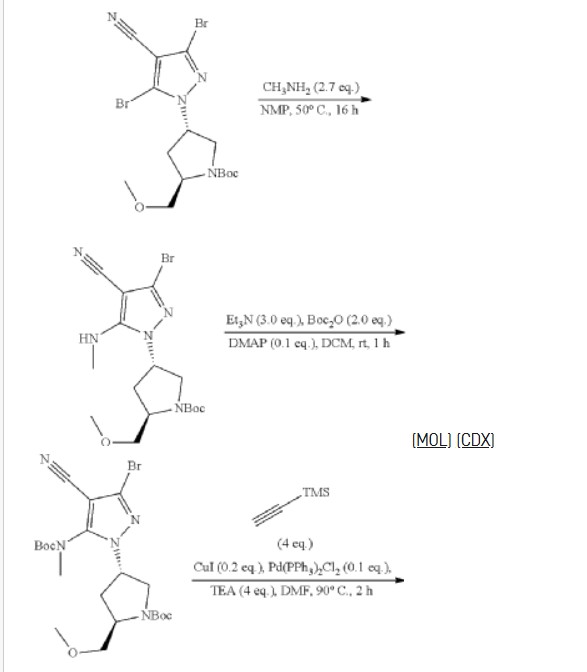
To a stirred solution of 3.5-dibromo-1H-pyrazole-4-carbonitrile (2.00 g, 7.97 mmol), tert-butyl (2R,4R)-4-hydroxy-2-(methoxymethyl)pyrrolidine-1-carboxylate (1.84 g, 7.97 mmol) and triphenylphosphine (3.14 g, 11.95 mmol) in THF (40.00 mL) was added diisopropyi azodicarboxylate (2.42 g, 11.95 mmol) dropwise at 0° C. under argon atmosphere. The reaction mixture was stirred for 2 h at room temperature. The resulting mixture was diluted with water (300 mL). The resulting mixture was extracted with EtOAc (3×200 mL). The combined organic layers was dried over anhydrous Na 2SO 4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with PE/EtOAc (5/1). The fractions contained desired product were combined and concentrated to afford tert-butyl (2R)-4-(3,5-dibromo-4-cyanopyrazol-1-yl)-2-(methoxymethyl)pyrrolidine-1-carboxylate (3.50 g, 94%) as a dark yellow solid. MS ESI calculated for C 15H 20Br 2N 4O 3 [M+H−100] +, 361.99, 363.99, 365.99; found 362.10, 364.10, 366.10.
To a stirred solution of tert-butyl (2R)-4-(3,5-dibromo-4-cyanopyrazol-1-yl)-2-(methoxymethyl)pyrrolidine-1-carboxylate (1.00 g, 2.15 mmol) in NMP (10.00 mL) was added CH 3NH 2(2.98 mL, 5.96 mmol) at room temperature under nitrogen atmosphere. The reaction mixture was stirred for 16 h at 50° C. under nitrogen atmosphere. The resulting mixture was diluted with water (30 mL) and extracted with EA (3×50 mL). The combined organic layers was washed with brine (3×20 mL), dried over anhydrous Na 2SO 4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with PE/EA (3/1). The fractions contained desired product were combined and concentrated to afford tert-butyl (2S,4R)-4-[3-bromo-4-cyano-5-(methylamino)pyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate (0.67 g, 35%) as an off-white solid. MS ESI calculated for C 16H 24BrN 5O 3 [M+H−100] +, 314.11, 316.11, found 314.10, 316.10.
To a stirred solution of tert-butyl (2R,4S)-4-[3-bromo-4-cyano-5-(methylamino)pyrazol-1-yl]-2-(methoxymethyl)pyrrollidine-1-carboxylate (20.30 g, 49.00 mmol) in DCM (300.00 mL) were added Boc 2O (20.97 mL, 98.01 mmol), DMAP (0.60 g, 4.90 mmol) and Et 3N (20.43 mL, 0.14 mol) at 0° C. under nitrogen atmosphere. The reaction mixture was stirred for 1 h at room temperature. The resulting mixture was diluted with water (3×200 mL) and extracted with DCM (3×200 mL). The combined organic layers was washed with brine (3×100 mL). The organic layer was dried over anhydrous Na 2SO 4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with PE/EA (1/1). The fractions contained desired product were combined and concentrated to afford (2R,4R)-4-[3-bromo-5-[(tert-butoxycarbonyl)(methyl)amino]-4-cyanopyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate (24.00 g, 95%) as an off-white solid. MS ESI calculated for C 21H 32BrN 5O 5[M+H] +, 514.16, 516.16, found 514.15, 516.15; 1H NMR (400 MHz, CDCl 3) δ 4.94-4.90 (m, 1H), 4.23-4.19 (m, 1H), 3.75-3.66 (m, 3H), 3.44-3.40 (m, 1H), 3.36 (s, 3H), 3.25 (s, 3H), 2.62-2.58 (m, 1H), 2.41-2.19 (m, 1H), 1.48 (s, 18H).
To a stirred mixture of (2R,4S)-4-[3-bromo-5-[(tert-butoxycarbonyl)(methyl)amino]-4-cyanopyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate (24.00 g, 46.65 mmol), CuI (1.78 g, 9.33 mmol), Pd(PPh 3) 2Cl 2 (3,27 g, 4.67 mmol) and trimethylsilylacetylene (19.78 mL, 0.20 mol) in DMF (240.00 mL) was added TEA (19,45 mL, 0.19 inol). The reaction mixture was degassed with nitrogen for three times and stirred for 2 h at 90° C. The resulting mixture was concentrated under reduced pressure. The residue was diluted with water (500 mL) and extracted with EA (4×500 mL). The combined organic layers was washed with brine (2×500 mL), dried over anhydrous Na 2SO 4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with PE/EA (2/1). The fractions contained desired product were combined and concentrated to afford tert-butyl (2R, 4S)-4-[5-[(tert-butoxycarbonyl)(methyl)amino]-4-cyano-3-[2-(trimethylisilyl)ethynyl]pyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate (2.4.00 g, 96%) as a brown solid. MS ESI calculated for C 26H 41N 5O 5Si [M+H]+, 532.29, found 532.40; 1H NMR (400 MHz, CDCl 3) δ 4.93-4.89 (m, 1H), 4.23-4.17 (m, 1H), 3.68-3.52 (m, 3H), 3.42-3.37 (m, 1H), 3.35-3.33 (m, 3H), 3.25-3.20 (m, 3H), 2.63-2.58 (m, 1H), 2.33-2.13 (m, 1H), 1.46 (s, 18H), 0.27 (s, 9H).
To a stirred solution of tert-butyl (2R,4S)-4-[5-[(tert-(butoxycarbonyl)(methyl)amino]-4-cyano-3-[2-(trimethylsilyl)ethynyl]pyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate (24.00 g, 45.14 mmol) in THF (200.00 mL) was added TBAF (67.70 mL, 67.70 mmol, 1 M in THF) at 0° C. The reaction mixture was stirred for 1 h at room temperature. The resulting mixture was concentrated under reduced pressure. The residue was diluted with water (500 mL) and extracted with EA (3×500 mL). The combined organic layers was washed with brine (2×500 mL), dried over anhydrous Na 2SO 4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with PE/EA (2/1). The fractions contained desired product were combined and concentrated to afford tert-butyl (2R,4S)-4-[5-[(tert-butoxycarbonyl)(methyl)amino]-4-cyano-3-ethynylpyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate (17.40 g, 83%) as an off-white solid, MS ESI calculated for C 23H 33N 5O 5 [M+H] +, 460.25, found 460.40; 1H NMR (400 MHz, CDCl 3) δ 4.93-4.89 (m, 1H), 4.22-4.18 (m, 1H), 3.84-3.46 (m, 3H), 3.42-3.37 (m, 1H), 3.37-3.31 (m, 4H), 3.24 (s, 3H), 2.62-2.59 (m, 1H), 2.28-2.24 (m, 1H), 1.46 (s, 18H).
To a stirred solution of ter tyl (2R,4S)-4-(5-[(tert-butoxycarbonyl)(methyl)amino]-4-cyano-3-ethynylpyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate (17.40 g, 37.86 mmol) in DMSO (30.00 mL) and EtOH (150.00 mL) were added 0.5 M NaOH (87.09 mL, 43.54 mmol) and H 2O 2(10.26 mL, 0.13 mol) at 0° C. The reaction mixture was stirred for 0.5 h at 0° C. Then the reaction mixture was warmed up to room temperature and stirred for another 0.5 h at room temperature. The resulting mixture was diluted with water (500 mL) and extracted with EA (3×500 mL). The combined organic layers was washed with brine (2×300 mL), dried over anhydrous Na 2SO 4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with PE/EA (1/2). The fractions contained desired product were combined and concentrated to afford tert-butyl (2R,4S)-4-[5-[(tert-butoxycarbonyl)(methyl)amino]-4-carbamoyl-3-ethynylpyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate (17.20 g, 95%) as an off-white solid. MS ESI calculated for C 23H 35N 5O 6 [M+H] +, 478.26, found 478.25; 1H NMR (300 MHz, CDCl 3) δ 6.80-6.74 (m, 1H), 5.69-5.62 (m, 1H), 5.04-5.00 (m, 1H), 4.23-4.19 (m, 1H), 3.75-3.67 (m, 3H), 3.49-3.42 (m, 1H), 3.39-3.32 (m, 3H), 3.14 (s, 3H), 2.72-2.60 (m, 1H), 2.32-2.21 (m, 1H), 1.62-1.31 (m, 18H).
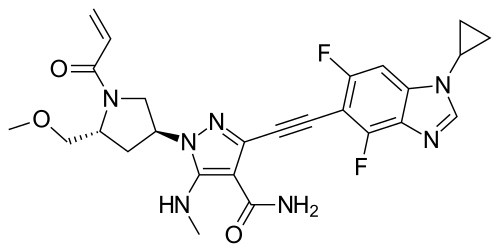
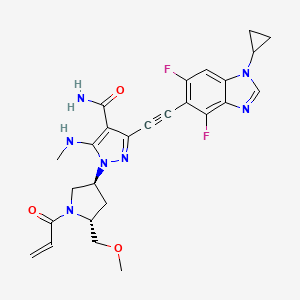
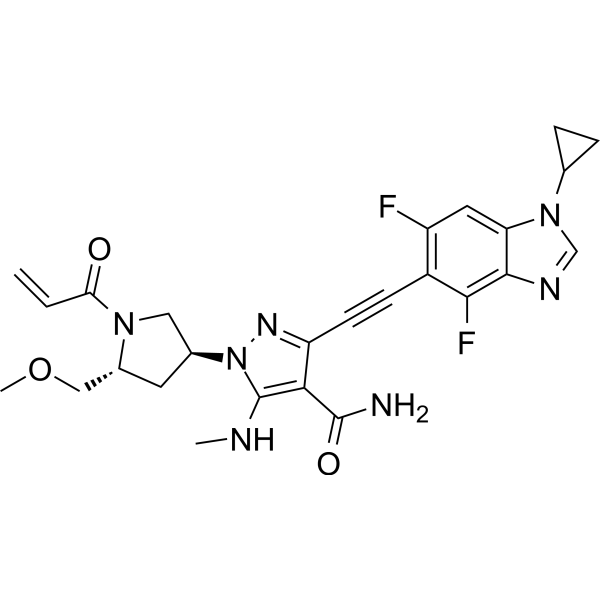
To a stirred mixture of tert-butyl (2R,4S)-4-[5-[(tert-butoxycarbonyl)(methyl)amino]-4-carbamoyl-3-ethynylpyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate (17.20 g, 36.02 minor) in DCM (170,00 mL) was added HCl (180.08 mL, 0.72 mol, 4 M in EA). The reaction mixture was stirred for 1 h at room temperature under argon atmosphere. The resulting mixture was concentrated and dried to afford 3-ethynyl-1-[(3S,5R)-5-(methoxymethyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide dihydrochloride (12.50 g, crude) as an off-white solid which was used in the next step directly without further purification. MS ESI calculated for C 13H 21Cl 2N 5O 2[M+H−2 HCl] +, 278.15, found 278.05.
To a stirred mixture of 3-ethynyl-1-[(3S,5R)-5-(methoxymethyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide dihydrochloride (12.50 g, 35.69 mmol) and K 2CO 3 (172 mL, 0.43 mol, 2.5 M) in THF (250.00 mL) was added acryloyl chloride (2.89 g, 32.15 mmol) dropwise at 0° C. under argon atmosphere. The reaction mixture was stirred for 10 min at 0° C. The resulting mixture was diluted with water (500 mL) and extracted with EA (3×500 mL). The combined organic layers was washed with brine (2×300 mL), dried over anhydrous Na 2SO 4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with DCM/MeOH (10/1). The fractions contained desired product were combined and concentrated to afford 3-ethynyl-1-[(3S,5R)-5-(methoxymethyl)-1-(prop-2-enoyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide (11.10 g, 84%) as an off-white solid. MS ESI calculated for C 16H 21N 5O 3[M+H] +, 332.16, found 332.20; 1H NMR (400 MHz, CDCl 3) δ 6.76 (s, 1H), 6.60-6.36 (m, 2H), 5.74-5.68 (m, 1H), 5.50-5.20 (m, 2H), 4.55-4.39 (m, 1H), 4.06-3.83 (m, 3H), 3.53-3.40 (m, 2H), 3.36-3.35 (m, 3H), 3.03-2.99 (m, 3H), 2.68-2.60 (m, 1H), 2.37-2.23 (m, 1H).
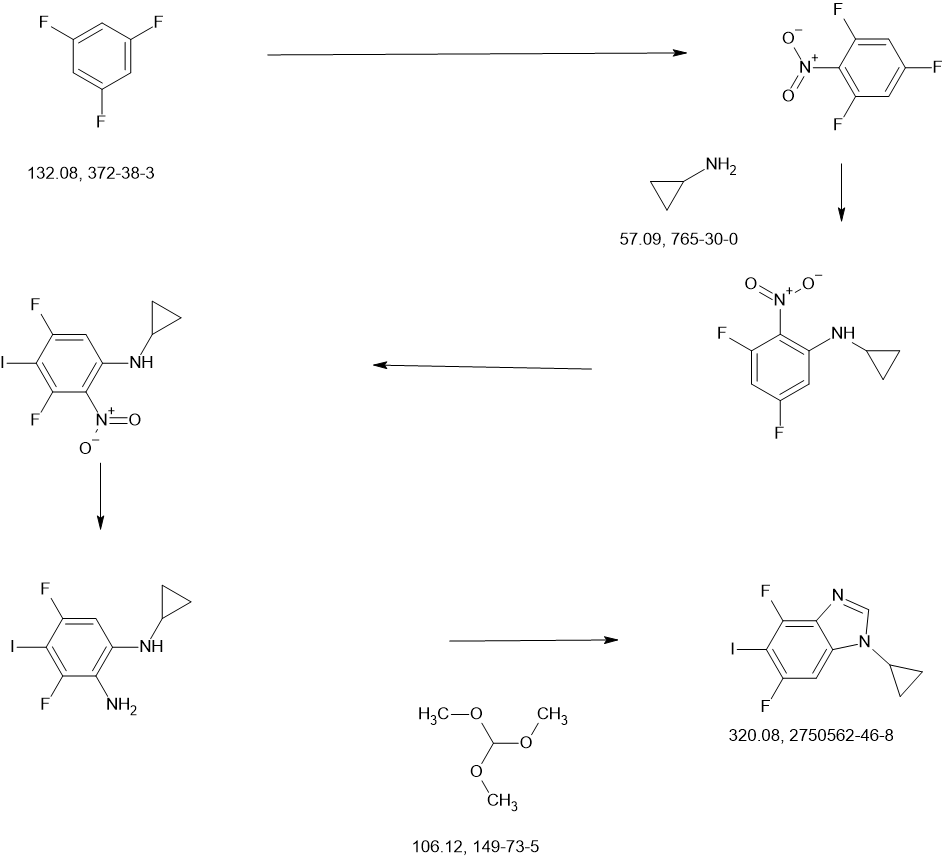
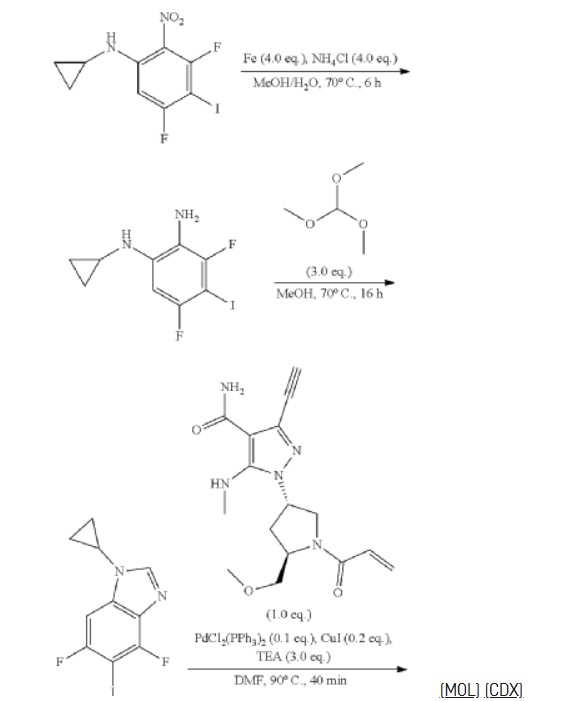
To a stirred solution of 1,3,5-trifluoro-2-nitrobenzene (4.50 g, 25.41 mmol) in EtOH (45.00 mL) was added aminocyclopropane (2.90 g, 50.82 mmol) dropwise at 0° C. under nitrogen atmosphere. The reaction mixture was stirred for 1 h at 0° C. The resulting mixture was diluted with water (100 mL) and extracted with EA (3×100 mL). The combined organic layers was washed with brine (2×50 mL), dried over anhydrous Na 2SO 4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with PE/EA (2/1). The fractions contained desired product were combined and concentrated to afford N-cyclopropyl-3,5-difluoro-2-nitroaniline (4.11 g, 85%) as a yellow solid. 1H NMR (400 MHz, CDCl 3) δ 7.64 (s, 6.80-6.74 (m, 1H), 6.30-6.27 (m, 1H), 2.59-2.54 (m, 1H), 1.01-0.83 (m, 2H), 0.76-0.61 (m, 2H).
To a stirred mixture of N-cyclopropyl-3,5-difluoro-2-nitroaniline (4.11 g, 19.19 mmol) in methanesulfonic acid (45.00 mL) was added NIS (4.53 g, 20.15 mmol) in portions at 0° C. The reaction mixture was stirred for 2 h at room temperature. The resulting mixture was quenched with ice/water (100 mL) at 0° C. The resulting mixture was basified to pH 8 with sat. NaOH and extracted with EA (3×100 mL). The combined organic layers was washed with brine (3×100 mL), dried over anhydrous Na 2SO 4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with PE/EA (1/1). The fractions contained desired product were combined and concentrated to afford N-cyclopropyl-3,5-difluoro-4-iodo-2-nitroaniline (4.50 g, 69%) a yellow solid. 1H NMR (400 MHz, CDCl 3) δ 7.53 (s, 1H), 6.90-6.88 (m, 1H), 2.57-2.54 (m, 1H), 1.07-0.85 (m, 2H ), 0.83-0.58 (m, 2H).
To a stirred mixture of N-cyclopropyl-3,5-difluoro-4-iodo-2-nitroaniline (4.40 g, 12.94 mmol) and NH 4Cl (2.77 g, 51.76 mmol) in EtOH (44.00 mL) and H 2O (8.80 mL) was added Fe (2.89 g, 51.76 mmol). The reaction mixtue was stirred at 70° C. for 6 h. The resulting mixture was diluted with water (150 mL) and extracted with EA×100 mL). The combined organic layers was washed with brine (2×50 mL), dried over anhydrous Na 2SO 4 and filtered. The filtrate was concentrated under reduced pressure to afford N-cyclopropyl-3,5-difluoro-4-iodobenzene-1,2-diamine (3.30 g, crude) as a brown oil which was used in the next step directly without further purification. MS ESI calculated for C 9H 9F 2IN 2 [M+H] +, 310.98, found 311.00; 1H NMR (300 MHz, CDCl 3) δ 6.68-6.61 (m, 1H), 2.50-2.41 (m, 1H), 0.89-0.73 (m, 2H), 0.78-0.51 (m, 2H).
To a stirred solution of N 1-cyclopropyl-3,5-difluoro-4-iodobenzene-1,2-diamine (3.30 g, 10.64 mmol) in MeOH (33.00 mL) was added trimethyl orthoformate (3.39 g, 31.92 mmol) at room temperature under nitrogen atmosphere. The reaction mixture was stirred for 16 h at 70° C. The resulting mixture was cooled down to room temperature and concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with PE/EA (1/2). The fractions contained desired product were combined and concentrated to afford 1-cyclopropyl-4,6-difluoro-5-iodo-1,3-benzodiazole (1.70 g, 50%) as a yellow solid. MS ESI calculated for C 11H 7F 2N 2 [M+H] +, 320.96, found 321.00; 1H NMR (300 MHz, CDCl 3) δ 7.91 (s, 1H), 7.22-7.18 (m, 1H), 3.41-3.36 (m, 1H), 1.31-1.02 (m, 4H).
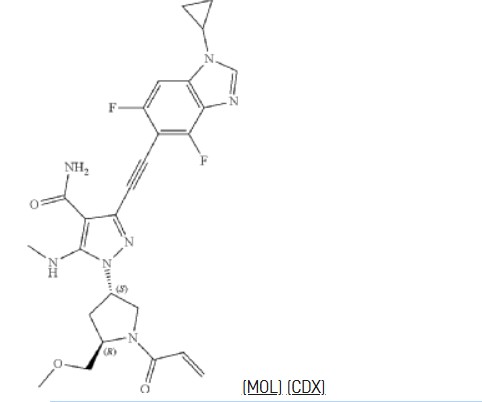
To a stirred solution of 1-cyclopropyl-4,6-difluoro-5-iodo-1,3-benzodiazole (0.97 g, 3.02 mmol), 3-ethynyl-1-[(3S,5R)-5-(methoxymethyl)-1-(prop-2-enoyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide (1.00 g, 3.02 mmol), Pd(PPh 3) 2Cl 2 (0.21 g, 0.30 mmol) and CuI (0.11 g, 0.60 mmol) in DMF (15.00 mL) was added TEA (0.92 g, 9.05 mmol) dropwise at room temperature. The reaction mixture was degassed with argon for three times and stirred for 40 min at 90° C. The resulting mixture was cooled down to room temperature and concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with DCM/MeOH (10/1) to afford the crude product. Then the crude product was further purified by reverse phase flash with the following conditions: column: C18 silica gel; mobile phase: ACN in water (10 mmol/L NH 3HCO 3), 10% to 50% gradient in 30 min; detector: UV 254 nm. The fractions contained desired product were combined and concentrated to afford 3-[2-(1-cyclopropyl-4,6-difluoro-1,3-b enzodiazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-(prop-2-enoyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide (0.51 g, 33%) as a white solid. MS ESI calculated for C 26H 27F 2N 7O 3 [M+H] +, 524.21, found 524.35; 1H NMR (300 MHz, CDCl 3) δ 7.99 (s, 1H), 7.30-7.12 (m, 2H), 6.83 (brs, EH), 6.67-6.32 (m, 2H), 5.84-5.73 (m, 1H), 5.64-5.12 (m, 2H), 4.71-4.38 (m, 1H), 4.25-3.84 (m, 3H), 3.60-3.33 (m, 5H), 3.20-3.08 (m, 3H), 2.86-2.70 (m, 1H), 2.37-2.31 (m, 1H), 1.40-0.89 (m, 4H).
PATENT
WO2021247969 Kinnate Biopharma Inc EG78
WO2023107980 solid state forms, Kinnate Biopharma Inc
Franovic A, Mohan A, Uryu S, Wu Q, Jiang P, Miller N, et al. (February 2022). “Activity of KIN-3248, a next-generation pan-FGFR inhibitor, against acquired FGFR-gatekeeper and molecular-brake drug resistance mutations”. Journal of Clinical Oncology. 40 (4_suppl): 461. doi:10.1200/JCO.2022.40.4_suppl.461.
Harding JJ, Perez CA, Kato S, Sharma M, Garmezy B, Quah CS, et al. (February 2023). “First in human (FIH) phase 1/1b study evaluating KIN-3248, a next-generation, irreversible pan-FGFR inhibitor (FGFRi), in patients (pts) with advanced cholangiocarcinoma (CCA) and other solid tumors harboring FGFR2 and/or FGFR3 gene alterations”. Journal of Clinical Oncology. 41 (4_suppl): TPS637-TPS637. doi:10.1200/JCO.2023.41.4_suppl.TPS637. S2CID256257314.

















{kind=link}