It's only fair to share...














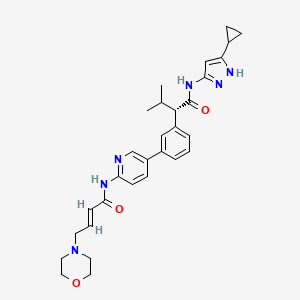
 Tacaciclib
2768774-66-7
AUR-102
Tacaciclib
2768774-66-7
AUR-102
- Tacaciclib
- SCHEMBL24548621
- GTPL12880
- 528.6 g/mol
- C30H36N6O3


- SAMAJDAR, Susanta
- PODDUTOORI, Ramulu
- PANDIT, Chetan
- MUKHERJEE, Subhendu
- GOSWAMI, Rajeev
Applicants
- AURIGENE ONCOLOGY LIMITED [IN]/[IN]

Inventors
WO2022229835
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022229835&_cid=P11-LR75VU-00171-1
Example- 1: Preparation of compound of formula (I)
Scheme-1: Preparation of KRM-A
- PODDUTOORI, Ramulu
- VIJAYKUMAR BHAT, Uday
- THIMMASANDRA SEETHAPPA, Devaraja
 Step-4
KRM-A 4
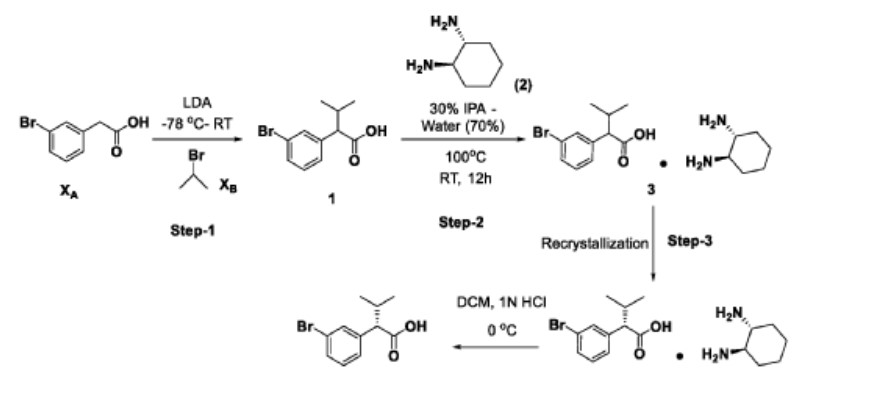
Step-1: Preparation of 2-(3-bromophenyl)-3-methylbutanoic acid (1)
2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3-bromophenyl) acetic acid (XA, 150 g, 0.69 mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mixture was stirred for 2h at -78 °C followed by a drop wise addition of isopropyl bromide (X B , 255 g, 2.07 mol) over a period of 30 min. The reaction mixture was stirred at room temperature overnight. Then, the reaction mixture was quenched with IN HC1 (pH 2) and the obtained product was extracted to ethyl acetate (500 mL x 3). The combined organic layer was washed with water followed by brine solution. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford the crude compound which was purified by silica column by eluting with 0-10% ethyl acetate-hexane system to afford the title compound (150 g, 83% yield) , HPLC purity-96%. The compound of formula (1) can also be prepared by the procedure described in CN 110590747.
Step-2: Preparation of Compound 3
2-(3-bromophenyl)-3-methylbutanoic acid (1, 510 g, 1.98 mol) was dissolved in 30% of IP A in water (10.2 L; 3.06 L of IPA-7.14 L of water) and ( 1L\ 2i ?)-cyclohexane-1,2-diamine (2, 113 g, 0.9 mol) was added. The reaction mixture was stirred at room temperature for 10 min until the precipitation was observed, then was heated to 100 °C until the solution became clear and stirred at the same temperature for another 30 min. The reaction mixture was allowed to slowly reach room temperature for 8-12h. The obtained solid was filtered and washed with 500 mL of 30% IPA-water mixture and dried under vacuum to afford the compound 3 (620 g, wet).
Work up (for Chiral purity): Small portion (100 mg) of compound 3 was taken in DCM (2-3 mL) and was added IN HC1 (pH 2) at 0 °C until the clear solution was observed. The compound was extracted into DCM, dried over NaiSCL and the solvent was evaporated to afford the title compound as white solid (20 mg). Chiral HPLC was recorded for this sample and 20.6% of undesired isomer was observed in chiral HPLC.
In order to improve the chiral purity of the title compound, the recrystallization method was performed as described below.
Step-3: Recrystallization
The compound 3 (619.90 g) was taken in 30% of IP A in water (12.4 L), then the mixture was heated to 100 °C until the solution became clear and was stirred at the same temperature for another 30min. The reaction mixture was allowed to reach room temperature slowly for 8-12h.
The obtained solid was filtered and washed with 500mL 30% IPA-water and dried under vacuum to afford a desired compound (360g, wet).
Work up for analysis (for Chiral purity): Small portion (100 mg) from above compound was taken in DCM (2-3mL), was added IN HC1 (pH 2) at 0 °C until the clear solution was observed and the compound was extracted to DCM, dried over NaiSCL and the solvent was evaporated to afford title compound as white solid (35 mg). Chiral HPLC was recorded for this sample and 10.3% of undesired isomer was observed in chiral HPLC.
The recrystallization method was repeated for three more times by using 30% of IPA in water as per the aforesaid procedure to obtain the purity of greater than 98.50% ee along with 0.27% other isomer to afford 286 g of compound 4.
Step-4: Preparation of (S)-2-(3-bromophenyl)-3-methylbutanoic acid (KRM-A)
The compound 4 (286 g) was taken in DCM (1.3 L), then was added IN HC1 at 0 °C until the clear solution was observed, and the compound was extracted to DCM (500 mL x 2). The organic layer was separated, washed with brine solution (500 mL) and dried over NaiSCL. The solvent was evaporated from the reaction mixture to afford title compound as white solid (148 g, 60% yield). Chiral HPLC: 98.50%
*H NMR (400MHz, DMSO-de): d 12.5 (s, 1H), 7.50-7.44 (m, 2H), 7.34-7.26 (m, 2H), 3.16 (d, 1H), 2.23-2.11 (m, 1H), 0.98 (d, 3H), 0.63 (d, 3H); Chiral HPLC: 98.50% retention time: 4,588 min.
Scheme-2: Preparation of compound of formula (I)
Step-4
KRM-A 4
Step-1: Preparation of 2-(3-bromophenyl)-3-methylbutanoic acid (1)
2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3-bromophenyl) acetic acid (XA, 150 g, 0.69 mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mixture was stirred for 2h at -78 °C followed by a drop wise addition of isopropyl bromide (X B , 255 g, 2.07 mol) over a period of 30 min. The reaction mixture was stirred at room temperature overnight. Then, the reaction mixture was quenched with IN HC1 (pH 2) and the obtained product was extracted to ethyl acetate (500 mL x 3). The combined organic layer was washed with water followed by brine solution. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford the crude compound which was purified by silica column by eluting with 0-10% ethyl acetate-hexane system to afford the title compound (150 g, 83% yield) , HPLC purity-96%. The compound of formula (1) can also be prepared by the procedure described in CN 110590747.
Step-2: Preparation of Compound 3
2-(3-bromophenyl)-3-methylbutanoic acid (1, 510 g, 1.98 mol) was dissolved in 30% of IP A in water (10.2 L; 3.06 L of IPA-7.14 L of water) and ( 1L\ 2i ?)-cyclohexane-1,2-diamine (2, 113 g, 0.9 mol) was added. The reaction mixture was stirred at room temperature for 10 min until the precipitation was observed, then was heated to 100 °C until the solution became clear and stirred at the same temperature for another 30 min. The reaction mixture was allowed to slowly reach room temperature for 8-12h. The obtained solid was filtered and washed with 500 mL of 30% IPA-water mixture and dried under vacuum to afford the compound 3 (620 g, wet).
Work up (for Chiral purity): Small portion (100 mg) of compound 3 was taken in DCM (2-3 mL) and was added IN HC1 (pH 2) at 0 °C until the clear solution was observed. The compound was extracted into DCM, dried over NaiSCL and the solvent was evaporated to afford the title compound as white solid (20 mg). Chiral HPLC was recorded for this sample and 20.6% of undesired isomer was observed in chiral HPLC.
In order to improve the chiral purity of the title compound, the recrystallization method was performed as described below.
Step-3: Recrystallization
The compound 3 (619.90 g) was taken in 30% of IP A in water (12.4 L), then the mixture was heated to 100 °C until the solution became clear and was stirred at the same temperature for another 30min. The reaction mixture was allowed to reach room temperature slowly for 8-12h.
The obtained solid was filtered and washed with 500mL 30% IPA-water and dried under vacuum to afford a desired compound (360g, wet).
Work up for analysis (for Chiral purity): Small portion (100 mg) from above compound was taken in DCM (2-3mL), was added IN HC1 (pH 2) at 0 °C until the clear solution was observed and the compound was extracted to DCM, dried over NaiSCL and the solvent was evaporated to afford title compound as white solid (35 mg). Chiral HPLC was recorded for this sample and 10.3% of undesired isomer was observed in chiral HPLC.
The recrystallization method was repeated for three more times by using 30% of IPA in water as per the aforesaid procedure to obtain the purity of greater than 98.50% ee along with 0.27% other isomer to afford 286 g of compound 4.
Step-4: Preparation of (S)-2-(3-bromophenyl)-3-methylbutanoic acid (KRM-A)
The compound 4 (286 g) was taken in DCM (1.3 L), then was added IN HC1 at 0 °C until the clear solution was observed, and the compound was extracted to DCM (500 mL x 2). The organic layer was separated, washed with brine solution (500 mL) and dried over NaiSCL. The solvent was evaporated from the reaction mixture to afford title compound as white solid (148 g, 60% yield). Chiral HPLC: 98.50%
*H NMR (400MHz, DMSO-de): d 12.5 (s, 1H), 7.50-7.44 (m, 2H), 7.34-7.26 (m, 2H), 3.16 (d, 1H), 2.23-2.11 (m, 1H), 0.98 (d, 3H), 0.63 (d, 3H); Chiral HPLC: 98.50% retention time: 4,588 min.
Scheme-2: Preparation of compound of formula (I)
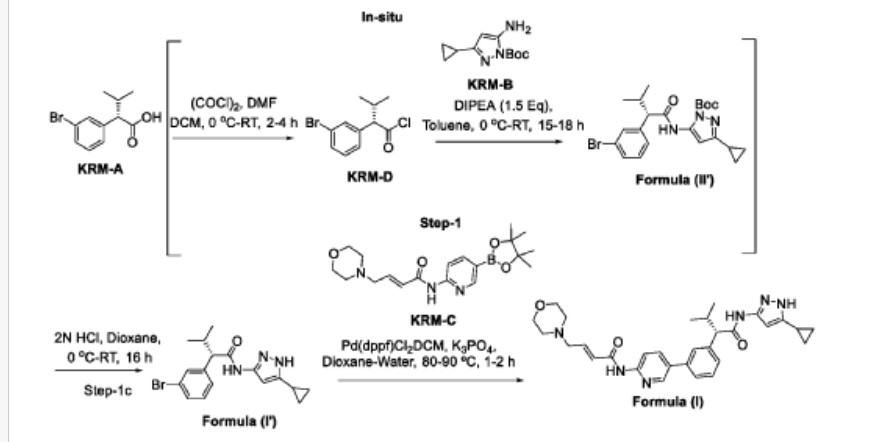 Step-1: Synthesis of (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-1H-pyrazol-3-yl)-3-methylbutanamide
Step-la: Preparation ofKRM-D
To a stirred solution of KRM-A (lOOg, O.388mol) in dry DCM (600 mL, 6 vol), a catalytic amount of DMF (10 mL) was added followed by oxalyl chloride (45 mL, 0.525 mol) dropwise at 0°C over a period of 30 min. After completion of addition, the reaction mixture was stirred for 15 min at the same temperature. The reaction mixture was allowed to reach room temperature and stirred for 2 to 4h. After completion of the reaction (reaction was monitored by TLC, acid chloride formation was checked by quenching an aliquot of reaction mixture with MeOH), the reaction mixture was concentrated under vacuum at 40°C-45°C to afford crude (S)- 2-(3-bromophenyl)-3-methylbutanoyl chloride (KRM-D). The crude KRM-D was dissolved in toluene (500mL) and used for next step.
Step-lb: Preparation of compound of formula (II)
(5)-2-(3-bromophcnyl)-3-mcthylbutanoyl chloride in toluene was added slowly to a pre-cooled solution (0 to 5 °C) of ieri-butyl 3-amino-5-cyclopropyl-1H-pyrazole- l-carboxylate (KRM-B, 95.5g, 0.427 mol) and N, N-diisopropylethyl amine (100 mL, 0.583 mol) in toluene (1.2 L) at 0 °C for the period of l-2h. The reaction mixture was allowed to reach RT and stirred overnight. The reaction mixture was then cooled to 0-5°C and washed with ice-cold 1.5N HC1 (3 x 500 mL). The organic layer was washed with sodium bicarbonate solution (500 mL), brine solution (500 mL), dried over anhydrous NaiSCL , filtered and concentrated under vacuum at 45-50°C to afford crude tert-butyl (S)-5-( 2-(3-bromophenyl)-3-methylbutanamido)-3-
cyclopropyl- lH-pyrazole- 1-carboxylate (compound of formula (IG)) as light brown oil (~180g, LCMS: m/z= 461.9 (M+H) + , HPLC: 80.80%, retention time:15.89 min) . The crude product was taken as such for next step without further purification.
Step-1 c: Preparation of compound of formula (I)
To a suspension of tert-butyl (S)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-1H-pyrazole-1-carboxylate (180 g, 1,731 mol) in dioxane (360 mL ) was added 2N aqueous HC1 (360 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature. After completion of the reaction, dioxane was concentrated, and the reaction mixture was diluted with water (500 mL) and basified with solid sodium bicarbonate (until pH-8). The obtained compound was extracted with DCM (700 mL x 3). The combined organic layers were washed with water (300 mL), brine solution (300 mL), and dried over anhydrous NaiSCL . The organic layer was concentrated to obtain a crude (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (Compound of formula (G)) as a semi-solid. The crude was dissolved in toluene (500 mL) and the solution was stirred for 18 h. The obtained solid was filtered and washed with toluene (100 mL) and n-heptane (200 mL). The solid was further dried under vacuum at 45-50°C for 6 h to afford a title compound (1 lOg, Yield: 78% over two steps). LCMS: m/z= 362 (M+H) + , HPLC: 97.66%, retention time: 24.10 min
Step-2: Preparation of (S, E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl) amino)-3-methyl-l-oxobutan-2- yl) phenyl) pyridin-2-yl)-4-morpholinobut-2-enamide (Compound of formula (I))
To a degassed solution of (5)-2-(3-bromophcnyl)-N-(5-cyclopropyl-1 H-pyrazol-3-yl)-3-methylbutanamide (50 g, 0.138 mol) and (E)-4 -morpholino-N-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)but-2-enamide (KRM-C, 56.6 g, 0.151 mol, 1.1 eq) (prepared according to the procedure described in W02020202001) in 1,4-dioxane (500 mL, 10 vol) and water (100 mL, 2 vol) was added K3PO4 tribasic (73.2 g, 0.345 mol, 2.5 eq) at room temperature The reaction mass was stirred for 20 min with argon purging (degassing). Pd(dppf)Ch.DCM (3.38 g, 0.0042 mol, and 0.03eq) was added to the reaction mixture and the reaction mixture was heated to 90°C for 1-2 h (The reaction was monitored by TLC using 10% methanol in DCM as solvent system).
After completion of the reaction, the reaction mass was cooled to room temperature and filtered through Celite ® bed. The bed was washed with 1, 4-dioxane (200 mL) and the filtrate was concentrated to get crude compound. The crude compound was dissolved in 5% methanol in DCM (400 mL) and washed with water (200 mL x 2). The aqueous layer was separated and
extracted with DCM (100 mL x 2). The combined organic layer was washed with brine solution, filtered and dried over sodium sulfate. The organic layer was concentrated under vacuum at 35-40°C to obtain crude title compound (~80g).
The crude compound of formula (I), (80 g) was dissolved in 700 mL of ethyl acetate. The reaction mixture was cooled to 15°C and 2N HC1 was slowly added (until pH ~1). The reaction mixture was then stirred at room temperature for 20 min and the layers were separated. The aqueous layer (containing the product) was washed with ethyl acetate (300 mL x 3). The aqueous layer was cooled to 0°C and adjusted the pH to ~8 using 20% aqueous NaiCCL solution. The product was extracted with 10% methanol in DCM (300 mL x 3). The combined organic layer was washed with water (300 mL), dried over sodium sulfate and filtered. The filtrate was treated with activated charcoal (16 g, 20% w/w with respect to crude input of 80 g), then the reaction mixture was stirred overnight at room temperature and filtered through Celite ® bed. The bed was washed with 5% methanol in DCM (~ 20 vol, until absence of product by TLC). The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound of formula (I) (70g, HPLC purity: 92.70%, retention time: 15.65 min).
Work-up for improved chiral purity: The above compound of formula (I) was dissolved in ethylacetate (~30 vol, 2L) and washed with aqueous citric acid (2 times, 400 mL x 1 and 200mL x 1), aqueous NaHCCL solution (2%, 500 mL x 1) and aqueous NaCl solution (10%, 500 mL x 1). The combined organic layer was dried over sodium sulfate and filtered. The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound of formula (I) (~60g).
*H NMR (400MHz, DMSO-rfe): d: 10.79 (s, 1H), 10.46 (s, 1H), 8.61 (d, 1H), 8.28 (d, 1H), 8.07-8.05 (m, 1H), 7.69 (s, 1H), 7.56 (d, 1H), 7.39 (m, 2H), 6.84-6.77 (m, 1H), 6.62 (s, 2H), 6.51 (d, 1H), 6.13 (s, 1H) , 3.62-3.59 (m, 4H), 3.35 (d, 1H), 3.15-3.13 (m, 2H), 2.42-2.39 (m, 5H), 1.80-1.77 (m, 1H), 0.98 (d, 3H) , 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.60 (m, 2H); LCMS: m/z= 529.25-free base (M+H) + , HPLC: 98.98%, retention time: 15.40 min.
Patent
WO-2022130304
PATENT
WO2023107861
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023107861&_cid=P11-LR76AB-05385-1
In some embodiments, the compound of formula (I) is (E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl)amino)-3-methyl-l-oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide or a pharmaceutically acceptable salt or a stereoisomer thereof (Compound 44).
Compound 44 is disclosed in WO 2016/193939 Al, published December 8, 2016, entitled “Substituted heterocyclyl derivatives as cdk inhibitors,” the entire contents of which are incorporated herein by reference. Compound 44A can be in the form of a fumaric acid salt or cocrystal as described in WO 2022/130304 Al, published June 23, 2022, entitled “Cocrystal of a cdk inhibitor,” the entire contents of which are incorporated herein by reference.
Example 3: Synthesis of Compounds 44A & 44B via Chiral Separation
Scheme-1
Step-1: Synthesis of (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-1H-pyrazol-3-yl)-3-methylbutanamide
Step-la: Preparation ofKRM-D
To a stirred solution of KRM-A (lOOg, O.388mol) in dry DCM (600 mL, 6 vol), a catalytic amount of DMF (10 mL) was added followed by oxalyl chloride (45 mL, 0.525 mol) dropwise at 0°C over a period of 30 min. After completion of addition, the reaction mixture was stirred for 15 min at the same temperature. The reaction mixture was allowed to reach room temperature and stirred for 2 to 4h. After completion of the reaction (reaction was monitored by TLC, acid chloride formation was checked by quenching an aliquot of reaction mixture with MeOH), the reaction mixture was concentrated under vacuum at 40°C-45°C to afford crude (S)- 2-(3-bromophenyl)-3-methylbutanoyl chloride (KRM-D). The crude KRM-D was dissolved in toluene (500mL) and used for next step.
Step-lb: Preparation of compound of formula (II)
(5)-2-(3-bromophcnyl)-3-mcthylbutanoyl chloride in toluene was added slowly to a pre-cooled solution (0 to 5 °C) of ieri-butyl 3-amino-5-cyclopropyl-1H-pyrazole- l-carboxylate (KRM-B, 95.5g, 0.427 mol) and N, N-diisopropylethyl amine (100 mL, 0.583 mol) in toluene (1.2 L) at 0 °C for the period of l-2h. The reaction mixture was allowed to reach RT and stirred overnight. The reaction mixture was then cooled to 0-5°C and washed with ice-cold 1.5N HC1 (3 x 500 mL). The organic layer was washed with sodium bicarbonate solution (500 mL), brine solution (500 mL), dried over anhydrous NaiSCL , filtered and concentrated under vacuum at 45-50°C to afford crude tert-butyl (S)-5-( 2-(3-bromophenyl)-3-methylbutanamido)-3-
cyclopropyl- lH-pyrazole- 1-carboxylate (compound of formula (IG)) as light brown oil (~180g, LCMS: m/z= 461.9 (M+H) + , HPLC: 80.80%, retention time:15.89 min) . The crude product was taken as such for next step without further purification.
Step-1 c: Preparation of compound of formula (I)
To a suspension of tert-butyl (S)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-1H-pyrazole-1-carboxylate (180 g, 1,731 mol) in dioxane (360 mL ) was added 2N aqueous HC1 (360 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature. After completion of the reaction, dioxane was concentrated, and the reaction mixture was diluted with water (500 mL) and basified with solid sodium bicarbonate (until pH-8). The obtained compound was extracted with DCM (700 mL x 3). The combined organic layers were washed with water (300 mL), brine solution (300 mL), and dried over anhydrous NaiSCL . The organic layer was concentrated to obtain a crude (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (Compound of formula (G)) as a semi-solid. The crude was dissolved in toluene (500 mL) and the solution was stirred for 18 h. The obtained solid was filtered and washed with toluene (100 mL) and n-heptane (200 mL). The solid was further dried under vacuum at 45-50°C for 6 h to afford a title compound (1 lOg, Yield: 78% over two steps). LCMS: m/z= 362 (M+H) + , HPLC: 97.66%, retention time: 24.10 min
Step-2: Preparation of (S, E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl) amino)-3-methyl-l-oxobutan-2- yl) phenyl) pyridin-2-yl)-4-morpholinobut-2-enamide (Compound of formula (I))
To a degassed solution of (5)-2-(3-bromophcnyl)-N-(5-cyclopropyl-1 H-pyrazol-3-yl)-3-methylbutanamide (50 g, 0.138 mol) and (E)-4 -morpholino-N-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)but-2-enamide (KRM-C, 56.6 g, 0.151 mol, 1.1 eq) (prepared according to the procedure described in W02020202001) in 1,4-dioxane (500 mL, 10 vol) and water (100 mL, 2 vol) was added K3PO4 tribasic (73.2 g, 0.345 mol, 2.5 eq) at room temperature The reaction mass was stirred for 20 min with argon purging (degassing). Pd(dppf)Ch.DCM (3.38 g, 0.0042 mol, and 0.03eq) was added to the reaction mixture and the reaction mixture was heated to 90°C for 1-2 h (The reaction was monitored by TLC using 10% methanol in DCM as solvent system).
After completion of the reaction, the reaction mass was cooled to room temperature and filtered through Celite ® bed. The bed was washed with 1, 4-dioxane (200 mL) and the filtrate was concentrated to get crude compound. The crude compound was dissolved in 5% methanol in DCM (400 mL) and washed with water (200 mL x 2). The aqueous layer was separated and
extracted with DCM (100 mL x 2). The combined organic layer was washed with brine solution, filtered and dried over sodium sulfate. The organic layer was concentrated under vacuum at 35-40°C to obtain crude title compound (~80g).
The crude compound of formula (I), (80 g) was dissolved in 700 mL of ethyl acetate. The reaction mixture was cooled to 15°C and 2N HC1 was slowly added (until pH ~1). The reaction mixture was then stirred at room temperature for 20 min and the layers were separated. The aqueous layer (containing the product) was washed with ethyl acetate (300 mL x 3). The aqueous layer was cooled to 0°C and adjusted the pH to ~8 using 20% aqueous NaiCCL solution. The product was extracted with 10% methanol in DCM (300 mL x 3). The combined organic layer was washed with water (300 mL), dried over sodium sulfate and filtered. The filtrate was treated with activated charcoal (16 g, 20% w/w with respect to crude input of 80 g), then the reaction mixture was stirred overnight at room temperature and filtered through Celite ® bed. The bed was washed with 5% methanol in DCM (~ 20 vol, until absence of product by TLC). The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound of formula (I) (70g, HPLC purity: 92.70%, retention time: 15.65 min).
Work-up for improved chiral purity: The above compound of formula (I) was dissolved in ethylacetate (~30 vol, 2L) and washed with aqueous citric acid (2 times, 400 mL x 1 and 200mL x 1), aqueous NaHCCL solution (2%, 500 mL x 1) and aqueous NaCl solution (10%, 500 mL x 1). The combined organic layer was dried over sodium sulfate and filtered. The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound of formula (I) (~60g).
*H NMR (400MHz, DMSO-rfe): d: 10.79 (s, 1H), 10.46 (s, 1H), 8.61 (d, 1H), 8.28 (d, 1H), 8.07-8.05 (m, 1H), 7.69 (s, 1H), 7.56 (d, 1H), 7.39 (m, 2H), 6.84-6.77 (m, 1H), 6.62 (s, 2H), 6.51 (d, 1H), 6.13 (s, 1H) , 3.62-3.59 (m, 4H), 3.35 (d, 1H), 3.15-3.13 (m, 2H), 2.42-2.39 (m, 5H), 1.80-1.77 (m, 1H), 0.98 (d, 3H) , 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.60 (m, 2H); LCMS: m/z= 529.25-free base (M+H) + , HPLC: 98.98%, retention time: 15.40 min.
Patent
WO-2022130304
PATENT
WO2023107861
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023107861&_cid=P11-LR76AB-05385-1
In some embodiments, the compound of formula (I) is (E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl)amino)-3-methyl-l-oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide or a pharmaceutically acceptable salt or a stereoisomer thereof (Compound 44).
Compound 44 is disclosed in WO 2016/193939 Al, published December 8, 2016, entitled “Substituted heterocyclyl derivatives as cdk inhibitors,” the entire contents of which are incorporated herein by reference. Compound 44A can be in the form of a fumaric acid salt or cocrystal as described in WO 2022/130304 Al, published June 23, 2022, entitled “Cocrystal of a cdk inhibitor,” the entire contents of which are incorporated herein by reference.
Example 3: Synthesis of Compounds 44A & 44B via Chiral Separation
Scheme-1
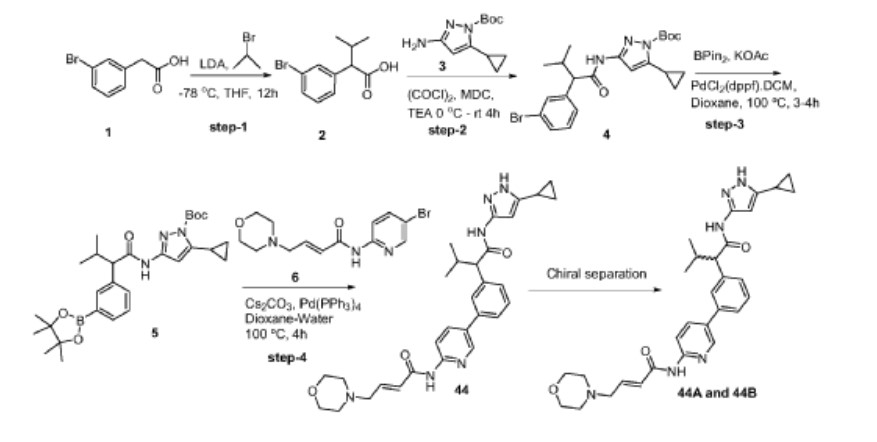 Step-1: Synthesis of 2-(3-bromophenyl)-3-methylbutanoic acid
[0352] 2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3 -bromophenyl) acetic acid (reagent-1, 150g, 0.69mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mass was stirred for 2h at -78 °C followed by the drop wise addition of Isopropyl bromide (255 g, 2.07mol) over a period of 30 min at -78 °C. The reaction mass was stirred at room temperature for overnight. The reaction mass was quenched with IN HC1 (pH 2) and product extracted to ethyl acetate (500mL x 3). The combined organic layer washed with water followed by brine, dried and concentrated under reduced pressure to afford the title crude compound which was purified by silica column by eluting with 0-10% ethyl acetate -hexane system to afford the title compound 2 (150g, 83% yield). LCMS: m/z = 254.80 (M-2H)’
Step-2: Synthesis of tert-butyl 3-(2-(3-bromophenyl)-3-methylbutanamido)-5-cyclopropyl-lH-pyrazole-1 -carboxylate
[0353] 2-(3-bromophenyl)-3-methylbutanoic acid (intermediate-2, 70g, 0.0.27mol) was dissolved in dry DCM (500 mL) and added oxalyl chloride (68 mL, 0.78mol) dropwise at 0 °C followed by addition of catalytic amount of DMF (0.8mL) and maintained reaction mass at same temperature for 30min. The reaction mass was allowed to room temperature and stirred for 4h, distilled off the solvent and excess oxalyl chloride under vacuum. Re-dissolved the residue in DCM (250 mL) and added slowly to the cooled solution of tert-butyl 3 -amino-5 -cyclopropyl- 1H-pyrazole-1 -carboxylate (intermediate-3, 49g, 0.218mol) and TEA (55 mL, 0.546mol) in THF (250 mL) at 0 °C for 30min, The reaction was stirred at room temperature for 12h then the reaction mass was concentrated under reduced pressure and the residue was dissolved in DCM, washed with saturated NaHCO3 solution and brine. The organic layer was dried over anhydrous sodium sulphate and concentrated under reduced pressure, the crude was purified by silica gel column chromatography by eluting with 15% ethyl acetate-hexane to afford the title compound 4 (90g, 71% ) LCMS: m/z = 363.80 (M-Boc+2).
Step-3: Synthesis of tert-butyl 5-cyclopropyl-3-(3-methyl-2-(3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)butanamido)-l H -pyr azole- 1 -carboxylate
[0354] To a degassed solution of tert-butyl 3-(2-(3-bromophenyl)-3-methylbutanamido)-5-cyclopropyl-lH-pyrazole-1 -carboxylate (intermediate-4, 90g, 0.193mol) and 4, 4, 4′, 4′, 5, 5, 5′, 5′-octamethyl-2,2′-bi(l,3,2-dioxaborolane) (62g, 0.25 Imol) in 1,4-Dioxane (500 mL) was added potassium acetate (37.80g, 0.386mol). The reaction mass was allowed to stir for 10 min with degassing at RT and added PdC12(dppf).DCM complex (12.5g, 0.015mol). The reaction mass was heated for 3-4 h at 100 °C. Reaction mixture cooled to RT and filtered on celite bed, filtrate evaporated to get dark brown liquid. The crude material was purified by silica column chromatography by eluting with 20% ethyl acetate in hexane to afford the compound 5 (90g, 86%). LCMS: m/z = 410 (M-Boc+1)+.
Step-4: Synthesis of (E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl)amino)-3-methyl-l-oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide
[0355] To a degassed solution of tert-butyl 5-cyclopropyl-3-(3-methyl-2-(3-(4, 4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)phenyl)butanamido)- 1 H-pyrazole- 1 -carboxylate, 5 (10g, 0.019mol) and (E)-N-(5-bromopyridin-2-yl)-4-morpholinobut-2-enamide (7.7g, 0.023mol) in
1,4-Dioxane (lOOmL) and water (40mL) followed by Cs2CO3 (14.5g, 0.045mol) were added. The reaction mass was allowed to stir for 10 min with degassing and added Pd(PPh3)4 (1.1g, 0.00095mol), heated the reaction mass for 4 h at 100 °C in a sealed tube. The reaction mass was cooled and diluted with brine solution. The aqueous layer was separated and re-extracted with ethyl acetate. The combined organic layer was evaporated to dryness and crude material was purified by silica column chromatography by eluting with 10%-l 5 % methanol in DCM to get desired pure compound 44 (4.5g, 44%). LCMS: m/z = 529.15 (M+H)+; HPLC: 95.17%, rt: 6.34 min.
[0356] Racemic (E)-N-(5 -(3 -( 1 -((5 -cyclopropyl- 1 H-pyrazol-3 -yl)amino)-3 -methyl- 1 -oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide was separated by using chiral preparative HPLC column (Method: Column: Chiral Pak IA (20mm X 250 mm, 5 micron), Elution: isocratic (50:50), A=ACN, B= MeOH, Flow: 20mL/min ) to afford the pure Isomer- 1 and Isomer-2.
Isomer-1 (Compound 44-A):
[0357] 1HNMR (DMSO-d6, 400MHz): 5 12.02 (s, 1H), 10.78 (s, 1H), 10.44 (s, 1H), 8.61 (s, 1H), 8.28 (d, 1H), 8.07-8.05 (m, 1H), 7.68 (s, 1H), 7.57 (d, 1H), 7.41-7.37 (m, 2H), 6.81-6.78 (m, 1H), 6.49 (d, 1H), 6.13 (s, 1H), 3.61-3.58 (m, 4H), 3.36-3.34 (m, 1H), 3.12 (d, 2H), 2.41-2.32 (m, 5H), 1.82-1.76 (m, 1H), 0.97 (d, 3H), 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.59 (m, 2H); LCMS: m/z = 529.15 (M+H)+; HPLC: 96.72%, rt: 6.39 min; Chiral HPLC: 97.68%, rt: 14.47.
Isomer-2 (Compound 44B):
[0358] 1HNMR (DMSO-d6, 400MHz): 5 12.02 (s, 1H), 10.78 (s, 1H), 10.44 (s, 1H), 8.61 (s, 1H), 8.28 (d, 1H), 8.07-8.04 (m, 1H), 7.68 (s, 1H), 7.57 (d, 1H), 7.41-7.37 (m, 2H), 6.81-6.78 (m, 1H), 6.50 (d, 1H), 6.14 (s, 1H), 3.61-3.58 (m, 4H), 3.36-3.34 (m, 1H), 3.12 (d, 2H), 2.40-2.39 (m, 5H), 1.82-1.76 (m, 1H), 0.97 (d, 3H), 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.60 (m, 2H); LCMS: m/z = 529.15 (M+H)+; HPLC: 96.24%, rt: 6.39 min; Chiral HPLC: 97.92%, rt: 8.80.
Example 4: Preparation of Compound 44-A via Chiral Synthesis
Preparation of KRM-A (chemical precursor to Compound 44-A)
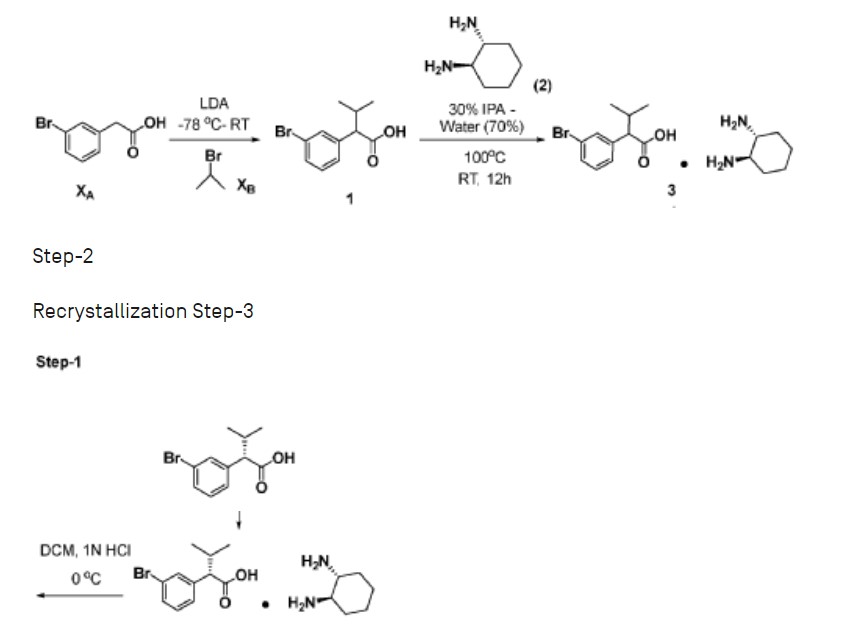
Step-1: Synthesis of 2-(3-bromophenyl)-3-methylbutanoic acid
[0352] 2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3 -bromophenyl) acetic acid (reagent-1, 150g, 0.69mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mass was stirred for 2h at -78 °C followed by the drop wise addition of Isopropyl bromide (255 g, 2.07mol) over a period of 30 min at -78 °C. The reaction mass was stirred at room temperature for overnight. The reaction mass was quenched with IN HC1 (pH 2) and product extracted to ethyl acetate (500mL x 3). The combined organic layer washed with water followed by brine, dried and concentrated under reduced pressure to afford the title crude compound which was purified by silica column by eluting with 0-10% ethyl acetate -hexane system to afford the title compound 2 (150g, 83% yield). LCMS: m/z = 254.80 (M-2H)’
Step-2: Synthesis of tert-butyl 3-(2-(3-bromophenyl)-3-methylbutanamido)-5-cyclopropyl-lH-pyrazole-1 -carboxylate
[0353] 2-(3-bromophenyl)-3-methylbutanoic acid (intermediate-2, 70g, 0.0.27mol) was dissolved in dry DCM (500 mL) and added oxalyl chloride (68 mL, 0.78mol) dropwise at 0 °C followed by addition of catalytic amount of DMF (0.8mL) and maintained reaction mass at same temperature for 30min. The reaction mass was allowed to room temperature and stirred for 4h, distilled off the solvent and excess oxalyl chloride under vacuum. Re-dissolved the residue in DCM (250 mL) and added slowly to the cooled solution of tert-butyl 3 -amino-5 -cyclopropyl- 1H-pyrazole-1 -carboxylate (intermediate-3, 49g, 0.218mol) and TEA (55 mL, 0.546mol) in THF (250 mL) at 0 °C for 30min, The reaction was stirred at room temperature for 12h then the reaction mass was concentrated under reduced pressure and the residue was dissolved in DCM, washed with saturated NaHCO3 solution and brine. The organic layer was dried over anhydrous sodium sulphate and concentrated under reduced pressure, the crude was purified by silica gel column chromatography by eluting with 15% ethyl acetate-hexane to afford the title compound 4 (90g, 71% ) LCMS: m/z = 363.80 (M-Boc+2).
Step-3: Synthesis of tert-butyl 5-cyclopropyl-3-(3-methyl-2-(3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)butanamido)-l H -pyr azole- 1 -carboxylate
[0354] To a degassed solution of tert-butyl 3-(2-(3-bromophenyl)-3-methylbutanamido)-5-cyclopropyl-lH-pyrazole-1 -carboxylate (intermediate-4, 90g, 0.193mol) and 4, 4, 4′, 4′, 5, 5, 5′, 5′-octamethyl-2,2′-bi(l,3,2-dioxaborolane) (62g, 0.25 Imol) in 1,4-Dioxane (500 mL) was added potassium acetate (37.80g, 0.386mol). The reaction mass was allowed to stir for 10 min with degassing at RT and added PdC12(dppf).DCM complex (12.5g, 0.015mol). The reaction mass was heated for 3-4 h at 100 °C. Reaction mixture cooled to RT and filtered on celite bed, filtrate evaporated to get dark brown liquid. The crude material was purified by silica column chromatography by eluting with 20% ethyl acetate in hexane to afford the compound 5 (90g, 86%). LCMS: m/z = 410 (M-Boc+1)+.
Step-4: Synthesis of (E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl)amino)-3-methyl-l-oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide
[0355] To a degassed solution of tert-butyl 5-cyclopropyl-3-(3-methyl-2-(3-(4, 4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)phenyl)butanamido)- 1 H-pyrazole- 1 -carboxylate, 5 (10g, 0.019mol) and (E)-N-(5-bromopyridin-2-yl)-4-morpholinobut-2-enamide (7.7g, 0.023mol) in
1,4-Dioxane (lOOmL) and water (40mL) followed by Cs2CO3 (14.5g, 0.045mol) were added. The reaction mass was allowed to stir for 10 min with degassing and added Pd(PPh3)4 (1.1g, 0.00095mol), heated the reaction mass for 4 h at 100 °C in a sealed tube. The reaction mass was cooled and diluted with brine solution. The aqueous layer was separated and re-extracted with ethyl acetate. The combined organic layer was evaporated to dryness and crude material was purified by silica column chromatography by eluting with 10%-l 5 % methanol in DCM to get desired pure compound 44 (4.5g, 44%). LCMS: m/z = 529.15 (M+H)+; HPLC: 95.17%, rt: 6.34 min.
[0356] Racemic (E)-N-(5 -(3 -( 1 -((5 -cyclopropyl- 1 H-pyrazol-3 -yl)amino)-3 -methyl- 1 -oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide was separated by using chiral preparative HPLC column (Method: Column: Chiral Pak IA (20mm X 250 mm, 5 micron), Elution: isocratic (50:50), A=ACN, B= MeOH, Flow: 20mL/min ) to afford the pure Isomer- 1 and Isomer-2.
Isomer-1 (Compound 44-A):
[0357] 1HNMR (DMSO-d6, 400MHz): 5 12.02 (s, 1H), 10.78 (s, 1H), 10.44 (s, 1H), 8.61 (s, 1H), 8.28 (d, 1H), 8.07-8.05 (m, 1H), 7.68 (s, 1H), 7.57 (d, 1H), 7.41-7.37 (m, 2H), 6.81-6.78 (m, 1H), 6.49 (d, 1H), 6.13 (s, 1H), 3.61-3.58 (m, 4H), 3.36-3.34 (m, 1H), 3.12 (d, 2H), 2.41-2.32 (m, 5H), 1.82-1.76 (m, 1H), 0.97 (d, 3H), 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.59 (m, 2H); LCMS: m/z = 529.15 (M+H)+; HPLC: 96.72%, rt: 6.39 min; Chiral HPLC: 97.68%, rt: 14.47.
Isomer-2 (Compound 44B):
[0358] 1HNMR (DMSO-d6, 400MHz): 5 12.02 (s, 1H), 10.78 (s, 1H), 10.44 (s, 1H), 8.61 (s, 1H), 8.28 (d, 1H), 8.07-8.04 (m, 1H), 7.68 (s, 1H), 7.57 (d, 1H), 7.41-7.37 (m, 2H), 6.81-6.78 (m, 1H), 6.50 (d, 1H), 6.14 (s, 1H), 3.61-3.58 (m, 4H), 3.36-3.34 (m, 1H), 3.12 (d, 2H), 2.40-2.39 (m, 5H), 1.82-1.76 (m, 1H), 0.97 (d, 3H), 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.60 (m, 2H); LCMS: m/z = 529.15 (M+H)+; HPLC: 96.24%, rt: 6.39 min; Chiral HPLC: 97.92%, rt: 8.80.
Example 4: Preparation of Compound 44-A via Chiral Synthesis
Preparation of KRM-A (chemical precursor to Compound 44-A)
 Step-4
KRM-A
Step-4
KRM-A 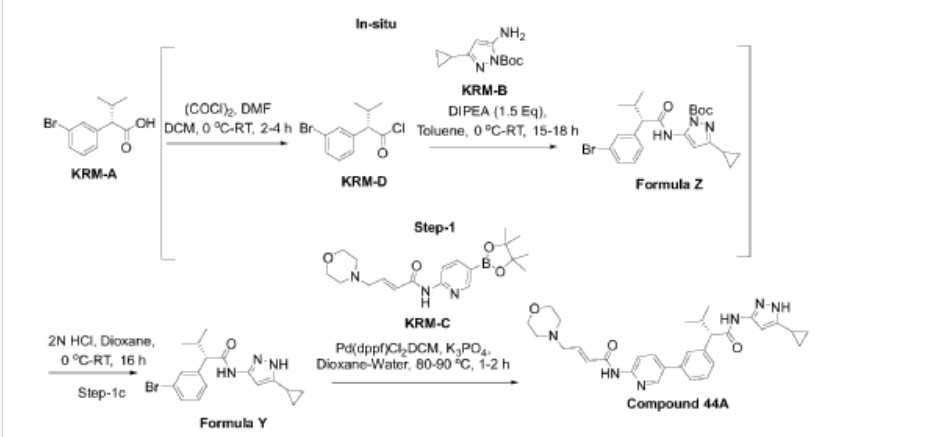 Step-1: Synthesis of (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylhutanamide
Step- la: Preparation of KRM-D
[0368] To a stirred solution of KRM-A (100g, 0.388mol) in dry DCM (600 mL, 6 vol), a catalytic amount of DMF (10 mL) was added followed by oxalyl chloride (45 mL, 0.525 mol) dropwise at 0 °C over a period of 30 min. After completion of addition, the reaction mixture was stirred for 15 min at the same temperature. The reaction mixture was allowed to reach room temperature and stirred for 2 to 4h. After completion of the reaction (reaction was monitored by TLC, acid chloride formation was checked by quenching an aliquot of reaction mixture with MeOH), the reaction mixture was concentrated under vacuum at 40°C-45°C to afford crude 2-(3-bromophenyl)-3 -methylbutanoyl chloride (KRM-D). The crude KRM-D was dissolved in toluene (500mL) and used for next step.
Step- lb: Preparation of compound of formula Z
[0369] (S)-2-(3-bromophenyl)-3 -methylbutanoyl chloride in toluene was added slowly to a pre-cooled solution (0 to 5 °C) of te/7-butyl 3 -amino-5 -cyclopropyl- IH-pyrazole-l -carboxylate (KRM-B, 95.5g, 0.427 mol) and N, N-diisopropylethyl amine (100 mL, 0.583 mol) in toluene (1.2 L) at 0 °C for the period of l-2h. The reaction mixture was allowed to attain RT and stirred for overnight. The reaction mixture was then cooled to 0-5°C and washed with ice-cold 1.5N HCI (3 x 500 mL). The organic layer was washed with sodium bicarbonate solution (500 mL),
brine solution (500 mL), dried over anhydrous Na2SO4, filtered and concentrated under vacuum at 45-50°C to afford crude tert-butyl (5)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-lH-pyrazole-1 -carboxylate (compound of formula Z) as light brown oil (~180g, LCMS: m/z= 461.9 (M+H)+, HPLC: 80.80%, retention time: 15.89 min). The crude product was taken as such for next step without further purification.
Step-lc: Preparation of compound of formula Y
[0370] To a suspension of tert-butyl (S)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-lH-pyrazole-1 -carboxylate (180 g, 1.731 mol) in dioxane (360 mL) was added 2N aqueous HC1 (360 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature.
[0371] After completion of the reaction, dioxane was concentrated, and the reaction mixture was diluted with water (500 mL) and basified with solid sodium bicarbonate (until pH-8). The resulted compound was extracted with DCM (700 mL x 3). The combined organic layers were washed with water (300 mL) and brine solution (300 mL), and dried over anhydrous Na2SO4. The organic layer was concentrated to get a crude (<S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (Compound of formula Y) as a semi solid. The crude was dissolved in toluene (500 mL) and the solution was stirred for 18 h. The solid formed was filtered and washed with toluene (100 mL) and n-heptane (200 mL). The solid was further dried under vacuum at 45-50°C for 6 h to afford a title compound (110g, Yield: 78% over two steps). LCMS: m/z= 362 (M+H)+, HPLC: 97.66%, retention time: 24.10 min
[0372] Step-2: Preparation of (S, E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl) amino)-3-methyl-l-oxobutan-2-yl) phenyl) pyridin-2-yl)-4-morpholinobut-2-enamide (Compound 44A)
[0373] To a degassed solution of (<S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (50 g, 0.138 mol) and (£)-4-morpholino-N-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)but-2-enamide (KRM-C, 56.6 g, 0.151 mol, 1.1 eq) (prepared according to the procedure described in W02020202001) in 1,4-dioxane (500 mL, 10 vol) and water (100 mL, 2 vol) was added K3PO4 tribasic (73.2 g, 0.345 mol, 2.5 eq) at room temperature The reaction mass was stirred for 20 min with argon purging (degassing). Pd(dppf)C12.DCM [l,l’-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with dichloromethane] (3.38 g, 0.0042 mol, and 0.03eq) was added and the reaction mixture was heated to 90°C for 1-2 h (The reaction was monitored by TLC using 10% methanol in DCM as solvent system).
[0374] After completion of the reaction, the reaction mass was cooled to room temperature and filtered through Celite® bed. The bed was washed with 1, 4-dioxane (200 mL) and the filtrate was concentrated to get crude compound. The crude compound was dissolved in 5% methanol in DCM (400 mL) and washed with water (200 mL x 2). The aqueous layer was separated and extracted with DCM (100 mL x 2). The combined organic layer was washed with brine solution, filtered and dried over sodium sulphate. The organic layer was concentrated under vacuum at 35-40°C to get crude title compound (~80g).
[0375] The crude compound 44A, (80 g) was dissolved in 700 mL of ethyl acetate. The reaction mixture was cooled to 15°C and 2N HC1 was slowly added (until pH ~1). The reaction mixture was then stirred at room temperature for 20 min and the layers were separated. The aqueous layer (containing the product) was washed with ethyl acetate (300 mL x 3). The aqueous layer was cooled to 0°C and adjusted the pH to ~8 using 20 % aqueous Na2COs solution. The product was extracted with 10% methanol in DCM (300 mL x 3). The combined organic layer was washed with water (300 mL), dried over sodium sulphate and filtered. The filtrate was treated with activated charcoal (16 g, 20% w/w with respect to crude input of 80 g), stirred overnight at room temperature and filtered through Celite® bed. The bed was washed with 5% methanol in DCM (~ 20 vol, till absence of product by TLC). The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound 44A (70g, HPLC purity: 92.70%, retention time: 15.65 min).
[0376] ‘ H NMR (400MHz, DMSO-^): <5: 10.79 (s, 1H), 10.46 (s, 1H), 8.61 (d, 1H), 8.28 (d, 1H), 8.07-8.05 (m, 1H), 7.69 (s, 1H), 7.56 (d, 1H), 7.39 (m, 2H), 6.84-6.77 (m, 1H), 6.62 (s, 2H), 6.51 (d, 1H), 6.13 (s, 1H), 3.62-3.59 (m, 4H), 3.35 (d, 1H), 3.15-3.13 (m, 2H), 2.42-2.39 (m, 5H), 1.80-1.77 (m, 1H), 0.98 (d, 3H), 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.60 (m, 2H);
LCMS: m/z= 529.25-free base (M+H)+, HPLC: 98.98%, retention time: 15.40 min.
Step-1: Synthesis of (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylhutanamide
Step- la: Preparation of KRM-D
[0368] To a stirred solution of KRM-A (100g, 0.388mol) in dry DCM (600 mL, 6 vol), a catalytic amount of DMF (10 mL) was added followed by oxalyl chloride (45 mL, 0.525 mol) dropwise at 0 °C over a period of 30 min. After completion of addition, the reaction mixture was stirred for 15 min at the same temperature. The reaction mixture was allowed to reach room temperature and stirred for 2 to 4h. After completion of the reaction (reaction was monitored by TLC, acid chloride formation was checked by quenching an aliquot of reaction mixture with MeOH), the reaction mixture was concentrated under vacuum at 40°C-45°C to afford crude 2-(3-bromophenyl)-3 -methylbutanoyl chloride (KRM-D). The crude KRM-D was dissolved in toluene (500mL) and used for next step.
Step- lb: Preparation of compound of formula Z
[0369] (S)-2-(3-bromophenyl)-3 -methylbutanoyl chloride in toluene was added slowly to a pre-cooled solution (0 to 5 °C) of te/7-butyl 3 -amino-5 -cyclopropyl- IH-pyrazole-l -carboxylate (KRM-B, 95.5g, 0.427 mol) and N, N-diisopropylethyl amine (100 mL, 0.583 mol) in toluene (1.2 L) at 0 °C for the period of l-2h. The reaction mixture was allowed to attain RT and stirred for overnight. The reaction mixture was then cooled to 0-5°C and washed with ice-cold 1.5N HCI (3 x 500 mL). The organic layer was washed with sodium bicarbonate solution (500 mL),
brine solution (500 mL), dried over anhydrous Na2SO4, filtered and concentrated under vacuum at 45-50°C to afford crude tert-butyl (5)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-lH-pyrazole-1 -carboxylate (compound of formula Z) as light brown oil (~180g, LCMS: m/z= 461.9 (M+H)+, HPLC: 80.80%, retention time: 15.89 min). The crude product was taken as such for next step without further purification.
Step-lc: Preparation of compound of formula Y
[0370] To a suspension of tert-butyl (S)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-lH-pyrazole-1 -carboxylate (180 g, 1.731 mol) in dioxane (360 mL) was added 2N aqueous HC1 (360 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature.
[0371] After completion of the reaction, dioxane was concentrated, and the reaction mixture was diluted with water (500 mL) and basified with solid sodium bicarbonate (until pH-8). The resulted compound was extracted with DCM (700 mL x 3). The combined organic layers were washed with water (300 mL) and brine solution (300 mL), and dried over anhydrous Na2SO4. The organic layer was concentrated to get a crude (<S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (Compound of formula Y) as a semi solid. The crude was dissolved in toluene (500 mL) and the solution was stirred for 18 h. The solid formed was filtered and washed with toluene (100 mL) and n-heptane (200 mL). The solid was further dried under vacuum at 45-50°C for 6 h to afford a title compound (110g, Yield: 78% over two steps). LCMS: m/z= 362 (M+H)+, HPLC: 97.66%, retention time: 24.10 min
[0372] Step-2: Preparation of (S, E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl) amino)-3-methyl-l-oxobutan-2-yl) phenyl) pyridin-2-yl)-4-morpholinobut-2-enamide (Compound 44A)
[0373] To a degassed solution of (<S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (50 g, 0.138 mol) and (£)-4-morpholino-N-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)but-2-enamide (KRM-C, 56.6 g, 0.151 mol, 1.1 eq) (prepared according to the procedure described in W02020202001) in 1,4-dioxane (500 mL, 10 vol) and water (100 mL, 2 vol) was added K3PO4 tribasic (73.2 g, 0.345 mol, 2.5 eq) at room temperature The reaction mass was stirred for 20 min with argon purging (degassing). Pd(dppf)C12.DCM [l,l’-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with dichloromethane] (3.38 g, 0.0042 mol, and 0.03eq) was added and the reaction mixture was heated to 90°C for 1-2 h (The reaction was monitored by TLC using 10% methanol in DCM as solvent system).
[0374] After completion of the reaction, the reaction mass was cooled to room temperature and filtered through Celite® bed. The bed was washed with 1, 4-dioxane (200 mL) and the filtrate was concentrated to get crude compound. The crude compound was dissolved in 5% methanol in DCM (400 mL) and washed with water (200 mL x 2). The aqueous layer was separated and extracted with DCM (100 mL x 2). The combined organic layer was washed with brine solution, filtered and dried over sodium sulphate. The organic layer was concentrated under vacuum at 35-40°C to get crude title compound (~80g).
[0375] The crude compound 44A, (80 g) was dissolved in 700 mL of ethyl acetate. The reaction mixture was cooled to 15°C and 2N HC1 was slowly added (until pH ~1). The reaction mixture was then stirred at room temperature for 20 min and the layers were separated. The aqueous layer (containing the product) was washed with ethyl acetate (300 mL x 3). The aqueous layer was cooled to 0°C and adjusted the pH to ~8 using 20 % aqueous Na2COs solution. The product was extracted with 10% methanol in DCM (300 mL x 3). The combined organic layer was washed with water (300 mL), dried over sodium sulphate and filtered. The filtrate was treated with activated charcoal (16 g, 20% w/w with respect to crude input of 80 g), stirred overnight at room temperature and filtered through Celite® bed. The bed was washed with 5% methanol in DCM (~ 20 vol, till absence of product by TLC). The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound 44A (70g, HPLC purity: 92.70%, retention time: 15.65 min).
[0376] ‘ H NMR (400MHz, DMSO-^): <5: 10.79 (s, 1H), 10.46 (s, 1H), 8.61 (d, 1H), 8.28 (d, 1H), 8.07-8.05 (m, 1H), 7.69 (s, 1H), 7.56 (d, 1H), 7.39 (m, 2H), 6.84-6.77 (m, 1H), 6.62 (s, 2H), 6.51 (d, 1H), 6.13 (s, 1H), 3.62-3.59 (m, 4H), 3.35 (d, 1H), 3.15-3.13 (m, 2H), 2.42-2.39 (m, 5H), 1.80-1.77 (m, 1H), 0.98 (d, 3H), 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.60 (m, 2H);
LCMS: m/z= 529.25-free base (M+H)+, HPLC: 98.98%, retention time: 15.40 min.


AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on ResearchgateRESEARCHGATE

Anthony Melvin Crasto Dr. | Facebook
join me on twitter Anthony Melvin Crasto Dr. | twitter +919321316780 call whatsaapp EMAIL. amcrasto@amcrasto ////////// REF POSTER SESSION: Molecular Targeted Agents| Volume 138, SUPPLEMENT 2, S47, October 01, 2020 //////Tacaciclib, GTPL12880, AUR-102 CC(C)C(C1=CC=CC(=C1)C2=CN=C(C=C2)NC(=O)C=CCN3CCOCC3)C(=O)NC4=NNC(=C4)C5CC5It's only fair to share...