Percent Composition: C 63.95%, H 6.90%, F 4.82%, O 24.34%
Literature References: Prepn: Bernstein et al.,J. Am. Chem. Soc.78, 5693 (1956); 81, 1689 (1959); Thoma et al.,ibid.79, 4818 (1957); Bernstein et al., Allen et al.,US2789118; US3021347 (1957, 1962, both to Am. Cyanamid). Comprehensive description: K. Florey, Anal. Profiles Drug Subs.1, 367-396, 423-442 (1972); D. H. Sieh, ibid.11, 593-614, 651-661 (1982).
Properties: Crystals, mp 269-271°. mp also reported as 260-262.5°. [a]D25 +75° (acetone). uv max: 238 nm (e 15800).
Melting point: mp 269-271°; mp also reported as 260-262.5°
Optical Rotation: [a]D25 +75° (acetone)
Absorption maximum: uv max: 238 nm (e 15800)
………………………………
Derivative Type: 16,21-Diacetate
CAS Registry Number: 67-78-7
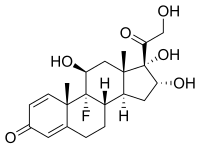
CAS Name: (11b,16a)-16,21-Bis(acetyloxy)-9-fluoro-11,17-dihydroxypregna-1,4-diene-3,20-dione
Percent Composition: C 66.34%, H 7.19%, F 4.37%, O 22.09%
Literature References: Prepd by stirring a suspension of triamcinolone in acetone in the presence of a trace of perchloric acid: Fried et al.,J. Am. Chem. Soc.80, 2338 (1958); Bernstein et al.,ibid.81, 1689 (1959); Bernstein, Allen, US2990401 (1961 to Am. Cyanamid). Alternate synthesis using 2,3-dibromo-5,6-dicyanoquinone: Hydorn, US3035050 (1962 to Olin Mathieson). Clinical trial in chronic asthma: I. L. Bernstein et al.,Chest81, 20 (1982). Comprehensive description: K. Florey, Anal. Profiles Drug Subs.1, 397-421 (1972); D. H. Sieh, ibid.11, 615-649 (1982).
Properties: Crystals, mp 292-294°. [a]D23 +109° (c = 0.75 in chloroform). uv max (abs alc.): 238 nm (e 14600). Sparingly sol in methanol, acetone, ethyl acetate.
Melting point: mp 292-294°
Optical Rotation: [a]D23 +109° (c = 0.75 in chloroform)
Absorption maximum: uv max (abs alc.): 238 nm (e 14600)
Percent Composition: C 67.40%, H 6.79%, F 3.05%, N 2.25%, O 20.52%
Literature References: Prepn: C. Cavazza et al.,DE2047218; eidem,US3749712 (1971, 1973 both to Sigma-Tau). Pharmacology: E. T. Ordonez, Arzneim.-Forsch.21, 248 (1971). Percutaneous absorption by rats and rabbits: W. H. Down et al.,Toxicol. Lett.1, 95 (1977). Clinical study: D. J. Tazelaar, J. Int. Med. Res.5, 338 (1977). HPLC analysis: S. Muck et al.,Boll. Chim. Farm.120, 240 (1981). For structure see Triamcinolone Acetonide.
Properties: Crystalline powder, mp 203-207°. [a]D20 +96 ±3° (c = 1 in ethanol). Sol in methanol, acetone, ethanol, dioxane, pyridine, DMF, chloroform. Insol in water.
Melting point: mp 203-207°
Optical Rotation: [a]D20 +96 ±3° (c = 1 in ethanol)
Percent Composition: C 67.65%, H 7.76%, F 3.57%, O 21.03%
Literature References: The hexacetonide ester of the potent glucocorticoid, triamcinolone, q.v. Prepn of syringeable suspension: Nash, Naeger, US3457348 (1969 to Am. Cyanamid). Anti-inflammatory activity in rabbits: I. M. Hunneyball, Agents Actions11, 490 (1981). Early clinical studies: Bilka, Minn. Med.50, 483 (1967); Layman, Peterson, ibid. 669. Clinical studies of intra-articular therapy in arthritis: R. C. Allen et al.,Arthritis Rheum.29, 997 (1986); M. Talke, Fortschr. Med.104, 742 (1986). Toxicity study: Tonelli, Steroids8, 857 (1966). Comprehensive description: V. Zbinovsky, G. P. Chrekian, Anal. Profiles Drug Subs.6, 579-595 (1977). For structure see Triamcinolone Acetonide.
Properties: Fine, white, needle-like crystals, mp 295-296° (dec), also reported as mp 271-272° (dec). uv max (ethanol): 238 nm (e 15500). [a]D25 +90±2° (c = 1.13% in chloroform). Soly in g/100 ml at 25°: chloroform and dimethylacetamide >5; ethyl acetate 0.77, methanol 0.59, diethyl carbonate 0.50, glycerin 0.42, propylene glycol 0.13; absolute alcohol 0.03; water 0.0004.
Triamcinolone was patented in 1956 and came into medical use in 1958.[8] It is available as a generic medication.[9] In 2019, it was the 107th most commonly prescribed medication in the United States, with more than 6million prescriptions.[10][11]
Skin is the layer of usually soft, flexible outer tissue covering the body of a vertebrate animal, with three main functions: protection, regulation, and sensation. Skin diseases are the medical condition that affects the skin, hair, nails and related muscle and glands.
Skin disorders vary greatly in symptoms and severity. They can be temporary or permanent, and may be painless or painful. Some have situational causes, while others may be genetic. Some skin conditions are minor, and others can be lifethreatening.
There are many different types of skin disorders which include rashes, dermatoses or skin eruptions. Such rashes, dermatoses or skin eruptions include acute, inflammatory reactions of the skin caused by an allergic or irritant reaction, other forms of eczema, lichen simplex chronicus. Chronic nature includes seborrheic dermatitis, psoriasis, and atopic dermatitis or caused by infection, irritation or aggravation of another condition such as occurs with acne, other rashes, dermatoses or skin eruptions, inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses, contact dermatitis, impetigo, urticarial and scabies.
Typical symptoms of the skin disorders include but not limited to raised bumps that are red or white, a rash, which might be painful or itchy, scaly or rough skin peeling skin, ulcers, open sores or lesions, dry, cracked skin, discolored patches of skin, fleshy bumps, warts, or other skin growths, changes in mole color or size a loss of skin pigment, excessive flushing or the like.
Atopic dermatitis (AD), also known as eczema or atopic eczema, is a type of inflammation of the skin (dermatitis). Atopic dermatitis (AD) is common worldwide. People of all ages from newborns to adults and older live with this condition. Symptoms range from excessively dry, itchy skin to painful, itchy rashes that cause sleepless nights and interfere with everyday life.
Topical corticosteroids have been the mainstay of treatment for atopic dermatitis over the past years, further the cure for atopic dermatitis involves Lifestyle modification, balanced diet intake, self-care measures, phototherapy, wet wrap therapy, use of medications like tacrolimus, pimecrolimus, crisaborole, dupilumab, ciclosporin, methotrexate, interferon gamma- lb, mycophenolate mofetil, and azathioprine or the like.
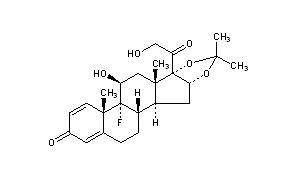
Triamcinolone Acetonide is a synthetic corticosteroid. Chemically it is [Pregna-1, 4-diene-3, 20-dione, 9-fluoro-l l, 21 -dihydroxy- 16, 17-[(1 methylethylidene) bis-(oxy)]-, (HP, 16a)-] with the empirical formula C24H31FO6 and molecular weight 434.50. Triamcinolone Acetonide is represented by compound of structural formula I
Triamcinolone Acetonide topical cream and ointment with strengths 0.025%, 0.1% and 0.5% (containing 0.25 mg/gm, 1 mg/gm & 5 mg/gm Triamcinolone Acetonide respectively) were approved in USA prior to Jan 1, 1982 under the trade name “Triamcinolone Acetonide” and were indicated for the relief of the inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses.
The commercially available products or product known in the prior art produces side effects such as burning, itching, irritation, or dryness of skin at site of application, folliculitis, hypertrichosis, acneiform eruptions, hypopigmentation, perioral dermatitis, allergic contact dermatitis, maceration of the skin, secondary infection, skin atrophy, striae and miliaria.
Pediatric patients may demonstrate greater susceptibility to topical triamcinolone -induced HPA axis suppression and Cushing’s syndrome than mature patients because of a larger skin surface area to body weight ratio. Hypothalamic -pituitary-adrenal (HPA) axis suppression, Cushing’s syndrome and intracranial hypertension have been reported in children receiving topical triamcinolone. Manifestations of adrenal suppression in children include linear growth retardation, delayed weight gain, low plasma cortisol levels, and absence of response to ACTH stimulation. Manifestations of intracranial hypertension include bulging fontanelles, headaches, and bilateral papilledema. Chronic corticosteroid therapy may interfere with the growth and development of children.
Making low dose compositions can present technical and economic challenges that are not present for higher dose formulations.
Examples
The following table 1 shows cream formulation containing lOO.OOmcg per gm, 50.00mcg per gm and 25.00mcg per gm of Triamcinolone Acetonide
Table – 1: cream
Drug Strength IQOmcg/gm 50mcg/gm 25mcg/gm
lOO.OOmcg per gm and for lOOgm, it is lO.OOmg*
50.00mcg per gm and for lOOgm, it is 5.00mg*
25.00mcg per gm and for lOOgm, it is 2.50mg**
Manufacturing process:
a) Dispensing following excipients – isopropyl myristate, glyceryl monostearate and white soft paraffin in vessel I;
b) Dispensing the following excipients – polysorbate 40 and purified water in vessel II;
c) Dispensing the following excipients methyl paraben, propylene glycol in vessel III; wherein methyl paraben is dissolved in propylene glycol to form a clear solution;
d) Dispensing the following active & excipients triamcinolone acetonide or salt thereof, propylene glycol in vessel IV; wherein triamcinolone acetonide or salt thereof is dissolved in propylene glycol to form clear solution;
e) Adding content of step (c) into content of step (b) and stirring to form uniform and homogeneous emulsion;
f) Heating content of step (b) and step (a) at about 75 °C and stirring to form a homogenous uniform emulsion;
g) Cooling the above emulsion gradually to temperature of about 25 °C – 30°C h) Adding the content of step (d) to the primary emulsion of (f) with constant stirring; and
i) Making up the volume of the emulsion with purified water to the required quantity.
Triamcinolone, 9a-fluoro-11b,16a,17,21-tetrahydroxypregna-1, 4-dien-3,20-dione (27.1.61), differs from dexamethsone in terms of chemical structure in that the a methyl group at C16 is replaced with a hydroxyl group. It is synthesized from the 21-O-acetate of hydrocortisone 27.1.17. In the first stage, both carbonyl groups of this compound undergo ketalization by ethylene glycol. Next, the hydroxyl group in the resulting diketal 27.1.53 is replaced with chlorine using thionyl chloride, and the product undergoes dehydrochlorination using an alkaline, during which the 21-O-acetyl group also is hydrolyzed. Acetylating the hydroxyl group once again with acetic anhydride gives a triene 27.1.54. Reacting this with osmium tetroxide gives the vicinal diol 27.1.55. The secondary hydroxyl group at C16 of this product undergoes acetylation by acetic anhydride in pyridine, which forms the diacetate 27.1.56. Treating the product with N-bromoacetamide in chloric acid gives a bromohydrin (27.1.57), which upon reaction with potassium acetate is transformed to an epoxide (27.1.58). Opening of the epoxide ring, using hydrofluoric acid, gives the corresponding 9-fluoro-11-hydroxy derivative 27.1.59. Upon microbiological dehydrogenation, the C1–C2 bond is oxidized to a double bond, forming triamcinolone acetate (27.1.60), the acetyl group of which is hydrolyzed, forming the desired triamcinolone (27.1.61) [30–32].
Triamcinolone is similar to dexamethasone in terms of pharmacological action, and it is better tolerated in some cases. Synonyms of this drug are ledercort, cenocort, delsolon, and others.
The synthesis of triamcinolone (23.2.1) starts from ketalization of cortisol 21-acetate (23.2.8) using ethylene glycol. Dehydration of the obtained compound (23.2.9) for creation of a double bond in position 16,17 of the steroid skeleton through the series of sequential reactions of chlorination, dehydrochlorination, hydrolysis, and acetylation produces 21-acetoxy-4,9(11),16-pregnatriene-3,20-dione (23.2.10), treatment of which with osmium tetroxide in benzene and pyridine produced diol (23.2.11), the secondary hydroxyl group of which, in position 16, was acetylated with acetic anhydride in pyridine to produce the diacetate (23.2.12). The obtained compound in dioxane and water was treated with N-bromoacetamide and 10% perchloric acid to yield bromohydrine (23.2.13). Dehydrobromination of the bromohydrine (23.2.13) with anhydrous potassium acetate in refluxing ethanol produced the epoxy-derivative (23.2.14). Opening of the epoxide ring in (23.2.14) with anhydrous hydrogen fluoride in chloroform produced (23.2.15). Microbiological dehydrogenation of the obtained product with Corynebacterium simplex produced crude diacetate (23.2.16), saponification of which produced triamcinolone (23.2.1) [108-110] (Scheme 23.7.).
Scheme 23.7. Synthesis of triamcinolone.
Triamcinolone is commonly used in the treatment of respiratory inflammation and improves airway reactivity, decreasing respiratory problems. Strangely, there are only few reviews of the pharmacotherapy of triamcinolone [111-113].
Glucocorticoids have a number of diverse effects in different body tissues. Glucocorticoids, in topical, oral and inhaled formulations, are useful for their anti-inflammatory and immunosuppressive properties. Several glucocorticoids such as budesonide and ciclesonide are used for treatment of several disorders.
The synthesis and purification of glucocorticoids have been disclosed at different instances. However, most of these synthetic procedures involve toxic solvents or long reaction times and are ineffective for large scale synthesis. For instance, US 3,92,9768 discloses a process for preparation of budesonide by reacting 16, 17-dihydroxy compound with aldehyde in solvents such as dioxane, methylene chloride or their combinations.
DE 4129535 discloses a process for the synthesis of Ciclesonide which involves the intermediate 16A, 17-[(7?,S)-cyclohexylmethylenedioxy]-l 13, 21-dihydroxy-pregna-l 4- dien-3,20-one which is obtained by an acid catalysed reaction of 11 , 16 , 17, 21-tetra hydroxypregna-l,4-dien-3,20-one with cyclohexane aldehyde.
WO 02/38584 discloses the synthesis of Ciclesonide by reacting corresponding 16, 17-ketals with a cyclohexane aldehyde in the presence of 70% perchloric acid, 1-nitropropane as solvent. However, perchloric acid is a dangerous solvent and can cause serious accidents with fatal consequences.
US Patent No. 6169178 relates to a process for the preparation of budesonide and of 16, 17- acetals of pregnane derivatives structurally co-related thereto comprising treating 16, 17-dios or of 16, 17-ketals or cyclic acetals with aldehydes in the presence of aqueous hydrobromic acid or hydroiodic acid used as reaction catalyst or solvents. However, hydroiodic and other hydrohalic solvents are corrosive, light sensitive and expensive. Further, these acids also post environmental problems. Notwithstanding the use of hydrohalo acids requires use of special equipment since they are extremely corrosive and consequently increase the cost of production.
US 5,55,6964 discloses a process for the preparation of budesonide by reacting 16 – Hydroxy Prednisolone in acetonitrile in the presence of /^-toluene sulfonic acid as a catalyst. There are certain other patents that use alkyl sulfonic acid instead of aryl sulfonic acid for the synthesis of budesonide or similar compounds. However, sulfonic acids are hazardous solvents and FDA has expressed significant concern over the presence or traces of sulfonic acid in pharmaceutical products. Hence, there is a need to have a process for the synthesis 16, 17- acetals of pregnane compounds that is industrially scalable and which does not involve the use of harmful solvents.
Example- 1: Process for preparation of 16-HPN from 3TR
Stage-I
Stage- 1 Stage-I I
Stage-IV
1 6-HPN acetate 1 6-HPN
Scheme 2: Synthesis of 16HPN from 3TR
Stage-I (oxidation)
Charge 750L of acetone (50 volume), 39L of purified water (2.60 volume) and 15 Kg of 3TR (40.93mol) in a SS Reactor at ambient temperature. Cool to -7°C to -5°C than added 6.0L of formic acid (159.03 mol) and 9.0 kg of potassium permanganate (56.95 mol). Maintain at – 5°Cto -3°C for 30 minutes. In-process check by TLC, 3TR should be less than 1.0%. Added 1.5kg sodium metabisulphite (7.89 mol solution in 12L of purified water at -5°C to -3°C then added 3.0 kg of hyflow super cell at 15°C (+2°C) and filter through 10.0 kg of hyflowbed at 27°C(+3°C) and wash with 150L of acetone Added 1.5 kg of activated charcoal, Stir and filter through hyflow bed and wash with 60L of acetone. Total filtrate was distilled under reduced pressure, while maintaining temperature below 45°C. Added 81L of purified water and cool to 5°C+5°C. Filter through centrifuge and wash with 156L of purified water. Wet material is dry at 60°+5°C till moisture less than 0.50%, Yield=15 kg, HPLC purity=98%.
Stage-II (Bromination)
Charge 75L of tetrahydrofuran, 16L of purified water and 15.0 kg of Stage-I (37.46 mol) in a glass reactor. Cool to -6°C (+2°C) and added 7.50 kg of dibromantin (26.23 mol) and 0.60L of perchloric acid (9.38 mol) and maintain at -6°C (+2°C) for one hour. In-process check by TLC, stage-I should be less than 0.50%. Reaction mass is quench in 390L of purified water at ~5°C. Raised the temperature to 25°C and maintained for 01 hour, filter through centrifuge and wash with 828L of purified water or till neutral pH. Wet material is dry at 40°C+5°C till moisture content should be less than 10%, Yield=21.0kg, HPLC purity=97%.
Stage-Ill (Debromination)
Charge 68.0L of N, N-dimethyl formamide(3.238volume) and 21.0kg of stage-II (42.22 mol) in glass reactor, start argon gas purging and cool to -5°C. Charge 13.0L of N,N- dimethylformamide (0.619volume) , 9.70L of dimethylsulfoxide(0.462volume), 1.62kg of chromium chloride hexahydrate (6.51 mol) and 1.94 kg of zinc dust (0.703 mol). Cool to – 10°Cand added 5.50L of thioglycolic acid (79.21 mol). Maintain for one hour while maintaining temperature around -10°C. In-process check by TLC, stage-II should be less than 1.0%. Added 310 L of purified water and cool to 0°C. Filter through centrifuge and wash with 1600L of purified water. Wet material is dry at 60°C+ (5°C) till moisture content less than 6.0%, Yield=15.0kg, HPLC Purity=90%.
Charge 150L of methylene chloride (10 volume), 150L of methanol (10 volume.) and 15.0kg (30.16 mol) of stage-Ill in a SS Reactor. Heat to clear solution then added 3.0 kg of activated charcoal (20%) and reflux for 04 hours, Filter through hyflow bed and wash with 75L of methylene chloride (5 volume), and 75L of methanol (5 volume) mixture. Total filtrate is distilled till last drop and added 75L (5 volume) of methylene dichloride, reflux for 04 hours than cool to 40°C+(5°C), Filter through centrifuge and wash with 15L (one volume) of methylene chloride. Wet material is dry at 60°C (+5°C) till moisture contents less than 1.0% (Yield =13.0kg, HPLC Purity=96%). Further charge 65.0L (5volume) of ethyl acetate and 13.0 kg (1.0 mol) of purified material. Heat to reflux and maintain for 04 hours under reflux, then cool to 40°C. Filter through centrifuge and wash with 13.0L (one volume) of ethyl acetate. Wet material is dry at 60°C (+5°C) till moisture contents less than 0.50%, Yield=12.0kg, HPLC Purity=98.6%.
Stage-IV (Deacetylation)
Charge 5.83L of methanol (10 volume) and 5.83L of methylene chloride (10 volume) in a glass flask and added 583 gm of 16-HPN acetate(1.397mol) at RT. Start argon gas purging and cool to 0°C to 5°C under argon purging. Prepare 11.66 gm of sodium hydroxide (0.2915mol) solution in 0.583L of methanol (one volume) under argon purging and cool to 0°Cto 5°C. Sodium hydroxide solution is charge in reaction mass at 0°C to 5°C. Maintained the reaction mass at 0°C to 5°C for one hour, In-process check by TLC against 16-HPN acetate it should be nil. Adjust pH to neutral by 21.40ml of acetic acid (0.3742 mol); distill under reduced pressure while maintaining temperature below 40°C, till dry. Cool to ambient temperature and added 1.166L of purified water (02 volume). Cool to 0°C and maintain for one hour. Filter and wash with 300ml of purified water. Dry at 60°C (+5°C) till moisture content less than 1.0%, Yield=490gm (93.50%), HPLC Purity=98.97%, Single impurity= 0.40%. Example 2: Process of synthesis of Budesonide from 16-HPN
16-HPN Budesonide
Charge 800 ml of aqueous hydrochloric acid (8 volume) in a glass flask, start nitrogen gas purging and Cool to -5°C and maintain for 15 min. then added 100 gm of stage-I (0.27 mol) at -5°C and stir for 15 min., added 30 ml of N-butyraldehyde (0.33 mol) while maintaining temperature -5°C to 0°C in around 30 minutes and maintain at 0°C to 5°C for 150 min. under stirring. In-process check by TLC against stage-I, it should be nil. Reaction mass is quench in 1200 ml of purified water (12 volume) at 5°C to 10°C and stir for 15 min. Added solution of 100 kg of sodium bicarbonate (1.19 mol) and 1 ml of purified water (10 volume) in reaction mass at 5°C to 10°C. Stir at 5°C to 10°C for 15 min. Filter and wash with purified water till neutral pH. Wet material is dry at 50°C (+5°C) till moisture contents less than 1.0 %, Yield =110 gm (96.49%), HPLC purity=96.45%, single impurity=1.29%, Epimer-A=47.76%, Epimer-B=49.69%.
(Purification)
Charge 2.5 L of methanol (25 volume) in a Glass flask and added 100 gm of above mentioned crude product. Dissolved at 25°C+5°C till clear solution, added 10 gm of activated charcoal and stir for 30 min. than filter through hyflow bed and wash with 200 ml of methanol (2 volume). Combined filtrates charged in a Glass flask and cool to 10°C to 15°C and added 5.40 L of purified water (54 volume) at 5°Cto 10°C, stir for 15min., filter and wash with purified water. Wet material is dry at 50°C (+5°C) under vacuum till moisture content less than 0.50%, Output=90.0gm, HPLC purity=99.66%, single impurity=0.1%, Epimer-A=44.47%, Epimer-B=55.01%.
Example 2.1: Scale-up process of manufacturing of Budesonide from 16-HPN
Charge 40 L of aqueous hydrochloric acid (8 volume) in a glass flask, start nitrogen gas purging and Cool to – 10°C and maintain for 15 min. then added 5.0 kg of stage-I (13.315 mol) at – 10°C and stir for 45 min. added 1.5 L of N-butyraldehyde (16.68 mol) while maintaining temperature -7°C to – 11°C in around 30 minutes and maintain at -2°C to -6°C for 60 min. under stirring In-process check by TLC against stage-I, it should be nil. Reaction mass is quench in 60 L of purified water (12 volume) at 5°C to 10°C and stir for 15 min. Added solution of 5.0 kg of sodium bicarbonate (59.525 mol) and 50L of purified water (10 volume) in reaction mass at 5°Cto 10°C. Stir at 5°C to 10°C for 15 min. Filter and wash with purified water till neutral pH. Wet material is dry at 50°C (+5°C) till moisture contents less than 1.0 %, Yield =5.293 kg (94.46%), HPLC purity=95.45%, single impurity=1.45%, Epimer-A=53.51 %, Epimer-B=43.78% Effect of temperature and its variation on epimer ratio (A and B) with respect to batch size (From lab to commercial batch)
Example 3: Process for synthesis ofCiclesonide from 16HPN
Preparation of cyclohexane carboxaldehydemetabisulphite complex
200gm of Cyclohexane carboxaldehyde (1.786 mol) was dissolved in 3.0L of denatured sprit (15 volume) and a solution of 190gm of sodium metabisulphite (1.827 mol) in 300ml of purified water (1.5 volume) was added. The resulting precipitate was filtered and washed with 1.0L of denatured sprit(5.0 volume) and dried under vacuum at 50°C, till moisture content less than 6.00%, Yield=400gm (97 %)
Stage I: Preparation of stage-I from 16-HPN
Cyclohexane carboxaldehyde
sodium metabisulphite complex
170gm of 16-HPN (0.4528 mol) was suspended in 3.40L of dichloromethane (20 volume) and treated with 340ml of 70% perchloric acid. (5.65 mol) and 110.5gm of cyclohexane carboxaldehyde metabisulphite complex (0.512 mol) was added in lots while maintaining the temperature between 0°Cto 5°C. The reaction mass was stirred at 0°C to 5°C for 03 hours. In- process check by TLC 16-HPN should be nil and then neutralized with 10% aqueous sodium bicarbonate solution. The organic layer was separated and concentrated under vacuum to obtain a residue which was stripped with methanol (1.0 volume). The solvent was concentrated and the residue was dissolved by refluxing in methanol (5.0 volume). The clear solution was cooled to 0°C to 5.0°C and the resulting solid was filtered and dried at 50°C till moisture content less than 0.50%, Yield=170.0gm (80.0%), HPLC purity=91.68%.
Stage -II Preparation of Ciclesonide from Stage -I
Stage-I Ciclesonide
158gm of stage-I (0.34mol) was suspended in 1.58L of methylene chloride (10.0 volume) at 25°C to 30°C. The reaction mass was chilled to 0°C to 5°C and 81.0ml of triethylamine(0.581 mol) was added, followed by the addition of 79.0ml of isobutyryl chloride [0.75 mol; diluted with 79.0 ml of methylene chloride (0.50 volume)] slowly at 0° to 5°C and maintained at same temperature for 60min. In-process check by TLC, Stage-I should be nil. The reaction mass was diluted with 2.53L of purified water (16.0 volume) , the organic layer was separated and washed with purified water till neutral pH, than organic layer was separated and concentrated under vacuum to obtained a residue. The residue was dissolved by refluxing in 948ml of methanol (6.0 volume); the clear solution was cooled to 0°C to 5°C under stirring and filtered. The product was dried under vacuum at ~50°C till moisture contents comes less than 0.50%, Yield=158.0 gm (87.0%), HPLC purity=95.74%.
(Purification)
120gm of Ciclesonide crude was dissolved by refluxing in 600ml of methanol. The clear solution was chilled to 20°C under stirring and filtered. The product was dried under vacuum at 90°C till moisture content less than 0.50%. Yield=105 gm (87.50%), HPLC purity=99.7 %.
Example 4: Process for synthesis of Desonide from 16HPN acetate
Stage-I : Preparation of Desonide acetate from 16 HPN acetate
Desonide acetate
16HPN acetate 190.0 ml of acetone (7.0 volume) was charged in a glass flask under nitrogen blanketing than added 27 gm of 16HPN acetate (0.0645mol) at ambient temperature. Temperature raised to 28°C (+2°C) and stir for 20 minutes. 1.35 ml of perchloric acid 70% (0.02 lmol) was added at 28°C (+2°C) and stir for 30 minutes. Temperature further raised to 35°C and stir for 60 minutes. In-process check by TLC against 16HPN acetate, it should be nil. Reaction mass cooled to 10°C, filtered and washed with purified water till neutral pH (~7) and finally washed with acetone. Wet material dried at 50°C+5°C till moisture content less than 0.50% to get stage-I. Yield =23gm (77.76%), HPLC Purity=98.28%
Stage-II: Preparation of Desonide from Desonide acetate
Desonide
Desonide acetate
200 ml of methanol (10 volume) and 200ml of methylene dichloride (10 volume) was charged in a glass flask and start argon gas purging. 20 gm of stage- 1st (0.0436mol) was added at ambient temperature. Cool to 0°C+5°C. 0.40gm of sodium hydroxide (O.Olmol) solution in 20ml of methanol (l.Ovolume) was added at 0°C+5°C. Stir at 0°C+5°C for 120 minutes. In-process check by TLC against stage- 1st it should be nil. Adjust pH to neutral (~7) by 2.0ml of acetic acid at 0°C+5°C. Distilled the solvent from reaction mass under vacuum while maintaining temperature below 40°C till the volume get reduced to 3 to 4 volume of the input. Cool to 0°C and further added 60ml of purified water and stir for 30 minutes. Filtered, washed with purified water till neutral pH (~7). Wet material dried at 50°C+5°C till moisture content less than 0.50% to get crude Desonide. Yield =14.70gm (80.92%), HPLC Purity=88.15%.
(Purification)
140 ml of methanol (10 volume) and 140 ml of methylene chloride (10 volume) was charged in a glass flask and added 14.0 gm of crude material (0.034mol) than stir till clear solution. Added 1.5 gm of activated charcoal and stir for 30 minutes than filtered through hyflow supercel bed and washed with 30ml of methanol and 30ml of methylene chloride mixture. Combined filtrate and distilled the solvent from reaction mass under vacuum while maintaining temperature below 40°C till the volume reduced to 3 to 4 volume of the input. Cool to 0°C. Filtered the reaction mass and washed with 10ml of precooled methanol. Wet material was dried at 50°C+5°C till moisture content less than 0.50% to get Desonide. Yield=8.60gm, HPLC Purity= 99.43%
lOOgm of 3TR (0.27 mol.)was suspended in 1300ml (13 volume) acetone. Cooled it to -5°C to -10°C than added 4.0 ml (0.062 mol.) perchloric acid solution and 50gm of dibromantin. Maintained the reaction at same temperature for 02 hours. In-process check by TLC against 3TR it should be nil. Added lOOgm of potassium carbonate solution (0.723 mol.) in 5 lots and reaction was maintained at 35°C+2°C. In-process check by TLC against step-I reaction mass, it should be nil. Cooled to 0°C (+5°C) and adjust pH neutral (~7) by 36ml of acetic acid (0.63 mol.). Added 3.0L of purified water (30 volume). Filter and washed with purified water till neutral pH (~7). Wet material was dried at 45°C (+2°C) till moisture content less than 0.50%. Yield =87gm, (83.36%), HPLC Purity=97.883%.
Stage – II:
80gm of stage-I (0.21 mol) was dissolved in 4.0L of acetone (50 volume) and 208ml of purified water (2.6 volume). Cool to -5°C (+2°C) than added 32ml of formic acid (0.85 mol.) and 48gm of potassium permagnate (0.30 mol.) at -5°C (+2°C). Reaction was maintained at – 5°C+2°Cfor one hour. In-process check by TLC against stage-I it should be nil. Added 8gm of sodium metabisulphite (0.042 mol.) In 80 ml purified water (01 volume) solution at -5°C (+2°C). Temperature raised up to 27°C and filtered through hyflow bed and washed with acetone. Acetone was distilled under vacuum till 3 to 4volume of stage-I than cool to 0°C to 5°C and added 480ml of purified water stir and filter and washed with purified water to get wet stage-II. Wet material was dried at 50°C (+5°C) till moisture content less than 3.0%. Yield =78.30gm, (89.88%), HPLC Purity=99.178%. Stage -III:
Stage-ll Stage-
300ml of hydrofluoric acid (12.60mol) was cooled at -25°C to -30°C than added 75gm of stage-II (0.180mol). Reaction was maintained at -25°C to -30°C for 04 hours. In-process check by TLC against stage-II, it should be nil. Reaction mass was cooled to -50°C than added 45ml of acetone (0.60volume) at -45°C to -50°C. Reaction was maintained at -45°C to -50°C for 02 hours. In-process check by TLC against before acetone reaction mass. Added 565ml of purified water at 0°C and 340ml of liq. ammonia at ~20°C than reaction mass was quenched in 410ml of liq. ammonia and 735ml of purified water solution at 15°C (+2°C), stir and filter and washed with purified water till neutral pH. Wet material was dried at 45°C to 50°C, Yield =78.50gm, (91.48%), HPLC Purity=91.593%.
(Purification)
76 gm of stage-Ill Crude (0.16 mol.) was dissolved in 760ml of methylene chloride (lOvolume) and 760ml of methanol (lOvolume) mixture at ambient temperature. Stir till clear solution and added 7.6gm of activated charcoal (0. lOvolume) than stir for 30minutes, filter through hyflow bed and washed with methanol (one volume) and methylene chloride (one volume) mixture. Total filtrate was distilled under vacuum till 3 to 4 volume of input. Cool to 0°C to 5°C and stir for 02 hours. Filtered and washed with minimum precooled methanol, Wet material was dried 45°C to 50°C till moisture contents less than 0.50%, Yield=62gm, HPLC Purity=98.633%.
Stage – IV (Process for synthesis of Triamcinolone acetonide from Stage – III):
Stage- Ill Triamcinolone acetonide
60gm of stage-Ill (0.13 mol) was dissolved in 600ml of methanol (lOvolume) and 600ml of methylene chloride (lOvolume) mixture under argon bubbling. Cool to -5°C+2°C and added 1.2gm of sodium hydroxide (0.03mol.) solution in 60ml of methanol (Olvolume) at -5°C (+2°C). Reaction maintained at -5°C (+2°C) for 03 hours. In-process check by TLC against stage-Ill, it should be nil. Adjust pH neutral (~7) by adding 1.8ml of acetic acid at -5°C (+2°C). Reaction mass was distilled at below 40°C under vacuum till 3 to 4 volume of input. Cool to 30°C and added 120ml of purified water, stir for one hour than filter and washed with purified water till neutral pH (~7). Wet material was dried at 45°C to 50°C till moisture content less than 0.50%, Yield =52gm, (95.04%), HPLC Purity=99.21%
(Purification)
50gm of crude material (0.12 mol.) was dissolved in 1100ml of acetone (22volume) and 100ml of purified water (02volume) at 50°C than added 2.5gm of activated charcoal and stir for one hour at same temperature, Filter through hyflow bed and washed with 120ml of acetone (2.40volume). Filtrate was distilled below 40°C under vacuum till 3 to 4 volume of input. Cool to 0°C to 5°Cand maintained for one hour at same temperature. Filter and washed with water. Wet material was dried at 45°C to 50°C till moisture content less than 0.50%, Yield=43gm, HPLC Purity=99.40%.
Example 6: Process for synthesis of Flunisolide from 16HPN acetate Stage -I (Preparation of Desonide acetate from 16HPN acetate):
1 6 H PN acetate eson e acetate
140ml of acetone (7 volume) was charged in glass flask and start argon blanketing than added 20 gm of 16-HPN acetate (0.048mol) at ambient temperature. Cooled to 28°C (+2°C). 1.0ml of perchloric acid 70% (0.016mol) was added at 28°C (+2°) C and stirred for 30 minutes. Temperature raised up to 35°Cand stirred for 60 minutes. In-process check by TLC against 16-HPN acetate, it should be nil. Reaction mass was cooled to 10°C (+2°C). Reaction mass was filtered and washed with purified water till neutral pH (~7) to get wet material. Wet material was dried at 50°C+5°C till moisture content less than 0.50% to get stage-lst. Yield=17.40gm, (79.40%), HPLC Purity=98.241%.
Stage -II (Preparation of Desonide from Desonide acetate):
170ml of methanol (lOvolume) and 170ml of methylene chloride (lOvolume) was charged in a glass flask and start inert atmosphere. 17gm of stage-lst (0.037mol) was added at ambient temperature. Cooled to -5°C. 0.4gm of sodium hydroxide (O.Olmol) solution in 17ml of methanol was added at 0°C (+5°C). Reaction mass was stirred for 02 hours at 0°C (+5°C). In- process check by TLC against stage- 1st it should be nil. Neutral pH (~7) was adjusted by acetic acid. Reaction mass was distilled under vacuum at below 40°C till ~ 100ml. Concentrated mass was cooled to 0°C (+5°C) and stir for one hour. Reaction mass was filtered and washed with precooled methanol to get wet material. Wet material was dried at 50°C (+5°C) till moisture content less than 0.50% to get stage-2nd. Yield=14.0gm, (90.67%), HPLC Purity=99.426%, Single impurity=0.136%.
Stage -III (Preparation of Flunisolide acetate from Desonide):
Desonide Flunisolide acetate
50ml of isopropenyl acetate (5 volume) was charged in a glass flask and added lOgm of stage-2nd (0.024mol) at ambient temperature than heated to 65°C and added 1.5ml of methane sulphonic acid (0.023mol) and temperature raised up to 80°C and stir for one hour. In-process check by TLC against stage-2, it should be nil. Reaction mass cooled to 25°C and adjust pH neutral (~7) by triethylamine. Reaction mass was distilled under vacuum till last drop and degases with acetonitrile. 90ml of acetonitrile (09 volume) was added and cooled to -5°C and than further added 10ml of purified water. lOgm of selectfluor(0.028mol) was added in two lots at 0°C(+5°C) in 02 volume of acetonitrile. Reaction mass was stirred at 10°C to 15°C for 12 hours. In-process check by TLC against before selectfluor reaction mass it should be nil. Adjust pH neutral (~7) by liq. ammonia solution at 0°C+5°C. Reaction mass was quenched in 500ml of purified water (lOOvolume) at ambient temperature. Reaction mass was filtered and washed with purified water till neutral pH (~7). Wet material was dried at 45°C+5°C till moisture content less than 0.50% to get stage-3rd. Yield=8.60gm, (75.17%), HPLC Purity= 94.12%.
Stage -IV (Preparation of Flunisolide from Flunisolide acetate):
Flunisolide acetate Flunisolide
80ml of methanol (lOvolume) and 80ml of methylene chloride (lOvolume) was charged in a glass flask under inert atmosphere at ambient temperature than added 8.0gm of stage-3r (0.017mol) at ambient temperature. Cooled to -5°C and added 0.16gm of sodium hydroxide (0.004mol) solution in 8ml of methanol at -5°C(+5°C) and stir for 02 hours at -5°C(+5°C). In-process check by TLC against stage-3 ‘ it should be nil. Adjust pH neutral(~7) by acetic acid and reaction mass was distilled under vacuum at below 40°C(+5°C) till ~40ml of volume. Cool to 0°C to 5°C and stir for one hour. Reaction mass was filtered and washed with precooled methanol to get wet material. Wet material was dried at 45°C (+5°C) till moisture content less than 0.50% to get Flunisolide crude. Yield=6.0gm, (82.30%), HPLC Purity=86.50%.
(Purification)
6.0gm of crude Flunisolide(0.014mol) was dissolved in 65ml of ethyl acetate (10.83volume) and 35ml of n-hexane (5.83volume) mixture and clear solution was passed through 60gm of silica gel column. Column was washed with 975ml of ethyl acetate (162.5volume) and 525ml of ft-hexane (87.5volume) mixture. Eluted fraction was distilled under vacuum till 3 to 4 volume of input than cooled it to 0°C and filter to get wet material. Wet material was dried at 50°C (+5°C) till moisture content less than 0.50% to get Flunisolide. Yield=4.28gm, HPLC Purity=95.60%.
Example 7: Process for synthesis of Triamcinolone from 3TR
S
lOOgm of 3TR (0.27mol) was suspended in 1300ml (13 volume) acetone. Cool to -5°C to- 10°C than added 4.0 ml (0.062mol) perchloric acid solution and 50gm of dibromantin. Reaction maintained at same temperature for 02 hours. In-process check by TLC against 3TR, it should be nil. Added lOOgm of potassium carbonate solution (0.723 mol) in 5 lots and reaction was maintained at 35°C (+2°C). In-process check by TLC against step-I reaction mass, it should be nil. Cool to 0°C+5°Cand adjust pH neutral (~7) by 36ml of acetic acid (0.63 mol). Added 3.0L of purified water (30 volume). Filter and washed with purified water till neutral pH (~7). Wet material was dried at 45°C (+2°C) till moisture content less than 0.50% to get stage-I. Yield=85.30gm, (81.74%), HPLC Purity=96.54%. Stage -II:
80gm of stage-I (0.21 mol) was dissolved in 4.0L of acetone (50 volume) and 208ml of purified water (2.6 volume). Cool to -5°C (+2°C) than added 32ml of formic acid (0.85 mol.) and 48gm of potassium per magnate (0.30 mol) at -5°C (+2°C). Reaction was maintained at same temperature for one hour. In-process check by TLC against stage-I, it should be nil. Added sodiummetabisulphite solution (8 gm in 80 ml of water) at -5°C+2°C. Temperature was raised up to 27°C and filtered through hyflow bed and washed with acetone. Acetone was distilled under vacuum till 3 to 4 volume of stage-I than further cooled to 0°C to 5°C and added 480ml of purified water, stirred, filter and washed with purified water to get wet stage- II. Wet material was dried at 50°C (+5°C) till moisture content less than 3.0% to get stage-II. Yield=82gm, (94.13%), HPLC Purity=97.75%.
Stage -III:
Stage-II Triamcinolone acetate
160ml of hydrofluoric acid (70%) (6.72mol) was cooled at -25°C to -30°C than added 40gm of stage-II (0.096mol). Reaction was maintained at -25°C to -30°C for 04 hours. In-process check by TLC against stage-II, it should be nil. Added 280ml of purified water at 0°C and 650ml of liq. ammonia at 20°C than reaction mass was quenched in 200ml of liq. ammonia and 500ml of purified water solution at 15°C(+2°C), stirred, filtered and washed with purified water till neutral pH(~7). Wet material was dried at 45°C to 50°C to get stage-Ill Yield=40gm, (95.42%), HPLC Purity=88.71%
(Purification)
40gm of stage-Ill crude (0.0916 mol) was refluxed in 160ml of acetone. Cool to 0°C. Filtered and washed with minimum precooled acetone. Wet material was dried at 50°C+5°C till moisture content comes less than 0.50% to get stage-Ill. Yield=24.9gm HPLC Purity=95.17%.
24gm of stage-Ill (0.055mol) was dissolved in 240ml of methanol (lOvolume) and 240ml of methylene chloride (lOvolume) mixture under argon bubbling. Cool to -5°C+2°C and added 0.48gm of sodium hydroxide (0.012mol) solution in 24ml of methanol (Olvolume) at – 5°C+2°C. Reaction was maintaining at -5°C (+2°C) for 03hours. In-process check by TLC against stage-Ill, it should be nil. Adjust pH neutral by adding 0.70ml of acetic acid at -5°C (+2°C). Reaction mass distilled at below 40°C under vacuum till 04-05 volume of input. Cooled to 0°C+5°Cand stir for one hour than filtered and washed with minimum precooled methanol. Wet material was dried at 45°C to 50°C till moisture content less than 0.50%. Yield=18.50gm, (85.29%), HPLC Purity=98.60%.
Example 8: Process for synthesis of Triamcinolone Hexacetonide from 3TR
S
lOOgm of 3TR (0.27288 mol) was suspended in 1300ml (13 volume) acetone. Cool to -5°C to -10°C than added 4.0 ml (0.0625 mol) perchloric acid solution and 50gm of dibromantin. Reaction was maintained at same temperature for 02 hours. In-process check by TLC against 3TR, it should be nil. Added lOOgm of potassium carbonate solution (0.723 mol) in 5 lots and reaction was maintained at 35°C (+2°C). In-process check by TLC against step-I reaction mass, it should be nil. Cool to 0°C (+5°C) and adjust pH neutral (~7) by 36ml of acetic acid (0.63 mol). Added 3.0L of purified water (30 volume). Filter and washed with purified water till neutral pH. Wet material was dried at45°C(+2°C) till moisture content less than 0.50% to get stage-lst. Yield =87gm, (83.36%), HPLC Purity=97.883%. Stage-II :
80gm of stage-I (0.21 mol) was dissolved in 4.0L of acetone (50 volume) and 208ml of purified water (2.6 volume). Cool to -5°C than added 32ml of formic acid (0.85 mol.) and 48gm of potassium permanganate (0.30 mol) at -5°C+2°C. Reaction maintained at -5°C (+2°C) for one hour. In-process check by TLC against stage-I, it should be nil. Added sodium metabisulphite solution (8 gm in 80 ml water) at -5°C (+2°C). Temperature raised up to 27°Cand filtered through hyflow bed and washed with acetone. Acetone was distilled under vacuum till 3 to 4 volume of stage-I than cooled to 0°C to 5°C and added 480ml of purified water, stirred, filtered and washed with purified water to get wet stage-II. Wet material was dried at 50°C (+5°C) till moisture content less than 3.0% to get stage-2nd. Yield=78.30gm, (89.88%), HPLC Purity=99.18%.
Stage – III:
300ml of hydrofluoric acid (12.60mol) was cooled at -25°C to -30°C than added 75gm of stage-II (0.180mol). Reaction was maintained at -25°C to -30°C for 04 hours. In-process check by TLC against stage-II. It should be nil. Reaction mass was cooled to -50°C than added 45ml of acetone (0.60volume) at -45°C to -50°C. Reaction maintained at -45°Cto – 50°C for 02 hours. In-process check by TLC against reaction input, it should be nil. Added 565ml of purified water at 0°C and 340ml of liq. ammonia at 20°C than reaction mass was quenched in 410ml of liq. ammonia and 735ml of purified water solution at 15°C(+2°C), stirred, filtered and washed with purified water till neutral pH (~7). Wet material was dried at 45°C to 50°Cto get stage-3rd. Yield=78.50gm, (91.48%), HPLC Purity=91.59%.
(Purification)
76 gm of stage-Ill Crude (0.16 mol) was dissolved in 760ml of methylene chloride (01 volume) and 760ml of methanol (lOvolume) mixture at ambient temperature. Stirred till clear solution and added 7.6gm of activated charcoal (0. lOvolume) than further stir for 30 minutes and filtered through hyflow bed and washed with methanol (one volume) and methylene chloride (one volume) mixture. Total filtrate was distilled under vacuum till 3 to 4 volume of input. Cooled to 0°C to 5°Cand stir for 02 hours. Filtered and washed with minimum precooled methanol. Wet material was dried at 45°C to 50°C till moisture content less than 0.50% to get purified stage-3rd. Yield=62gm, HPLC Purity=98.633%
Stage -IV : (Preparation of Triamcinolone acetonide from Stage – III)
Stage- Ill Triamcinolone acetonide
60gm of stage-Ill (0.1259 mol) dissolved in 600ml of methanol (lOvolume) and 600ml of methylene chloride (lOvolume) mixture under inert atmosphere. Cool to -5°C and added 1.2gm of sodium hydroxide (0.03mol) solution in 60ml of methanol (Olvolume) at -5°C (+2°C). Reaction maintained at -5°C+2°C for 03 hours. In-process check by TLC against stage-Ill, it should be nil. Adjust pH neutral (~7) by adding 1.8ml of acetic acid at -5°C+2°C. Reaction mass was distilled below 40°C under vacuum till 3 to 4 volume of input. Cool to
30°C and added 120ml of purified water, stir for one hour than filtered and washed with purified water till neutral pH (~7). Wet material was dried at 45°C to 50°C till moisture content less than 0.50% to get stage-4111 (Triamcinolone acetonide). Yield=52gm, (95.04%), HPLC Purity=99.21%.
(Purification)
50gm of crude material (0.12 mol) dissolved in 1100ml of acetone (22volume) and 100ml of purified water (02volume) at 50°C than added 2.5gm of activated charcoal and stirred for one hour at same temperature. Filter through hyflow bed and washed with 120ml acetone (2.40volume). Filtrate was distilled below 40°C under vacuum till 3 to 4 volume of input. Cool to 0°C to 5°C and maintained for one hour at same temperature. Filtered and washed with water. Wet material was dried at 45°C to 50°C till moisture content less than 0.50% to get purified stage-4th. Yield =43gm, HPLC Purity=99.40%
-V: (Preparation of Triamcinolone Hexacetonide from Triamcinolone acetonide):
50ml of pyridine (lOvolume) charged in a glass flask and added lOgm of Triamcinolone acetonide (0.023mol) at ambient temperature. Heated to 80°C to 90°C than added 10ml of 3, 3-dimethyl butyryl chloride (O.l lmol) at 80°C to 90°C. Stirred at 80°C to 90°C for 02 hours. In-process check by TLC against Triamcinolone acetonide, it should be nil. Reaction mass cooled to ambient temperature and reaction mass was quenched in 1000ml of purified water (lOOvolume) at ambient temperature, stir for one hour than filtered and washed with purified water till neutral pH (~7). Wet material was dried at 50°C (+5°C) till moisture content less than 1.0% to get stage-5th (Triamcinolone Hexacetonide). Yield=12gm, (97.90%), HPLC Purity=98.63%.
(Purification)
120ml of methanol and 120ml of methylene chloride charged in a glass flask and added 12gm of crude material, stir till clear solution than added 1.2gm of activated charcoal and stir for 30 minutes. Filtered through hyflow bed and washed with 12ml of methanol and 12ml of methylene chloride mixture. Total filtrate was distilled under vacuum at below 40°C till 5 to 6 volume of crude. Cooled to 0°C+5°C and stir for one hour. Filtered and washed with 12ml of precooled methanol. Wet material was dried at 40°C+5°C till moisture content less than 0.50% to get TrimcinolneHexacetonide. Yield=8.8gm, HPLC Purity=99.625%
//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
The derivativetriamcinolone acetonide is the active ingredient in various topical skin preparations (cream, lotion, ointment, aerosol spray) designed to treat skin conditions such as rash, inflammation, redness, or intense itching due to eczema[15] and dermatitis.[16]
Side effects of triamcinolone are similar to other corticoids. In short-term treatment up to ten days, it has very few adverse effects; however, sometimes gastrointestinal bleeding is seen, as well as acute infections (mainly viral) and impaired glucose tolerance.[4]
No acute overdosing of triamcinolone has been described.[17]
Interactions
Drug interactions are mainly pharmacodynamic, that is, they result from other drugs either adding to triamcinolone’s corticoid side effects or working against its desired effects. They include:[4][17]
Triamcinolone and other drugs can also influence each other’s concentrations in the body, amounting to pharmacokinetic interactions such as:[4][17]
Rifampicin, phenytoin, carbamazepine and other inducers of the liver enzyme CYP3A4[19] speed up metabolization of triamcinolone and can therefore reduce its effectiveness.
Conversely, CYP3A4 inhibitors such as ketoconazole and itraconazole can increase its concentrations in the body and the risk for adverse effects.
Blood concentrations of ciclosporin can be increased.
Triamcinolone is a glucocorticoid that is about five times as potent as cortisol, but has very little mineralocorticoid effects.[4]
Pharmacokinetics
When taken by mouth, the drug’s bioavailability is over 90%. It reaches highest concentrations in the blood plasma after one to two hours and is bound to plasma proteins to about 80%. The biological half-life from the plasma is 200 to 300 minutes; due to stable complexes of triamcinolone and its receptor in the intracellular fluid, the total half-life is significantly longer at about 36 hours.[4][5]
A small fraction of the substance is metabolized to 6-hydroxy- and 20-dihydro-triamcinolone; most of it probably undergoes glucuronidation, and a smaller part sulfation. Three quarters are excreted via the urine, and the rest via the faeces.[4][17]
Due to corticoids’ mechanism of action, the effects are delayed as compared to plasma concentrations. Depending on the route of administration and the treated condition, the onset of action can be from two hours up to one or two days after application; and the drug can act much longer than its elimination half-life would suggest.[4][5]
Chemistry
Triamcinolone is a syntheticpregnanecorticosteroid and derivative of cortisol (hydrocortisone) and is also known as 1-dehydro-9α-fluoro-16α-hydroxyhydrocortisone or 9α-fluoro-16α-hydroxyprednisolone as well as 9α-fluoro-11β,16α,17α,21-tetrahydroxypregna-1,4-diene-3,20-dione.[20][21]
The substance is a light-sensitive, white to off-white, crystalline powder, or has the form of colourless, matted crystals. It has no odour or is nearly odourless. Information on the melting point varies, partly due to the substance’s polymorphism: 260 to 263 °C (500 to 505 °F), 264 to 268 °C (507 to 514 °F), or 269 to 271 °C (516 to 520 °F) can be found in the literature.[4]
In 2010, TEVA and Perrigo launched the first generic inhalable triamcinolone.[22]
According to Chang et al. (2014), “Triamcinolone acetonide (TA) is classified as an S9 glucocorticoid in the 2014 Prohibited List published by the World Anti-Doping Agency, which caused it to be prohibited in international athletic competition when administered orally, intravenously, intramuscularly or rectally”.[23]
See also
Glucocorticoid (a chart comparing various glucocorticoids)



















































![{\displaystyle [\alpha ]_{D}^{20}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b67b300e0d318e964fcdab92c872ed89d1d4f43f)

