Antineoplastic
RP-5152; RP-5237; PI3K delta inhibitors (cancer), Rhizen/Incozen; PI3K delta inhibitors (B-cell lymphoma/hematological cancers), Incozen/Rhizen; TGR-1202; TG-1202; RV-1001; umbralisib tosylate; umbralisib; RP-5264; RP-5307; dual PI3Kdelta/CK1 inhibitor (cancer), TG Therapeutics; Ukoniq
Umbralisib (TGR-1202) is an orally available PI3K delta inhibitor, targeting the delta isoform with nanomolar potency and several fold selectivity over the alpha, beta, and gamma isoforms of PI3K. The delta isoform of PI3K is strongly expressed in cells of hematopoietic origin and is believed to be important in the proliferation and survival of B-cell lymphocytes. Inhibition of PI3K delta signaling with umbralisib has demonstrated robust activity in numerous pre-clinical models and primary cells from patients with hematologic malignancies. Umbralisib is currently in Phase 3 clinical development in combination with Ublituximab for patients with hematologic malignancies.
Umbralisib, sold under the brand name Ukoniq, is a medication for the treatment of marginal zone lymphoma (MZL) and follicular lymphoma (FL).[2] It is taken by mouth.[2]
The most common side effects include increased creatinine, diarrhea-colitis, fatigue, nausea, neutropenia, transaminase elevation, musculoskeletal pain, anemia, thrombocytopenia, upper respiratory tract infection, vomiting, abdominal pain, decreased appetite, and rash.[2]
Umbralisib is a kinase inhibitor including PI3K-delta and casein kinase CK1-epsilon.[2][3][4] Umbralisib was approved for medical use in the United States in February 2021.[2][5]
In April 2019, the FDA granted umbralisib Orphan drug designations for the treatment of nodal MZL, extranodal MZL, and splenic MZL. In January 2019, the FDA granted Breakthrough Therapy Designation for the treatment of MZL in patients who had received at least one prior anti-CD20 regimen, based on the interim data from the MZL umbralisib monotherapy cohort in the UNITY-NHL study. In March 2020, the drug was granted Orphan status for treatment of FL By June 2019, the confirmation of registration path to submit umbralisib for accelerated approval was obtained from the MZL cohort of the UNITY-NHL Phase IIb trial .
In August 2020, the FDA accepted the NDA for review; the MZL indication (patients with previously treated MZL who have received at least one prior anti-CD20 based regimen) was accepted for Priority Review with a PDUFA date of February 15, 2021, while the FL indication (patients with previously treated FL who have received at least two prior systemic therapies) was accepted for standard review with a PDUFA date of June 15, 2021.
In February 2021, the drug was granted accelerated approval by the FDA for second-line MZL and for fourth-line FL, based on results of UNITY-NHL. At that time, commercial launch was expected in the coming days
Medical uses
Umbralisib is indicated for adults with relapsed or refractory marginal zone lymphoma (MZL) who have received at least one prior anti-CD20-based regimen; and adults with relapsed or refractory follicular lymphoma (FL) who have received at least three prior lines of systemic therapy.[2][1]
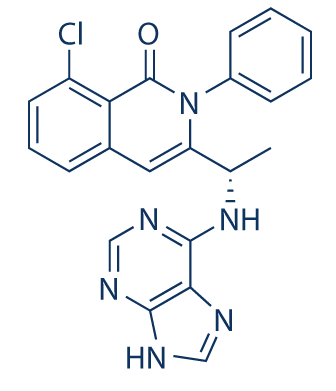
Umbralisib is a kinase inhibitor. The active pharmaceutical ingredient is umbralisib tosylate with the molecular formula C38H32F3N5O6S and a molecular weight of 743.75 g/mol. The chemical name for umbralisib tosylate is (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo [3, 4-d] pyrimidin-1-yl)-ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4- methylbenzenesulfonate and has the following structure:
Umbralisib tosylate is white to light brown powder that is freely soluble in dimethyl sulfoxide, soluble in methanol, and practically insoluble in water. The ionization constant (pKa) of umbralisib tosylate is 2.71.
UKONIQ tablets are for oral administration. Each tablet contains 200 mg of umbralisib free base equivalent to 260.2 mg of umbralisib tosylate. The tablets also contain inactive ingredients: croscarmellose sodium, hydroxypropyl betadex, hydroxypropyl cellulose, magnesium stearate and microcrystalline cellulose.
The tablet coating film consists of FD&C Blue No. 1, FD&C Yellow No. 5, ferric oxide yellow, hypromellose 2910, polydextrose, polyethylene glycol 8000, titanium dioxide and triacetin.
Indications & Dosage
INDICATIONS
Marginal Zone Lymphoma
UKONIQ is indicated for the treatment of adult patients with relapsed or refractory marginal zone lymphoma (MZL) who have received at least one prior anti-CD20-based regimen.
This indication is approved under accelerated approval based on overall response rate [see Clinical Studies]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Follicular Lymphoma
UKONIQ is indicated for the treatment of adult patients with relapsed or refractory follicular lymphoma (FL) who have received at least three prior lines of systemic therapy.
This indication is approved under accelerated approval based on overall response rate [see Clinical Studies]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Adverse effects
The prescribing information provides warnings and precautions for adverse reactions including infections, neutropenia, diarrhea and non-infectious colitis, hepatotoxicity, and severe cutaneous reactions.[2]
History
It has undergone clinical studies for chronic lymphocytic leukemia (CLL).[6][7] Three year data (including follicular lymphoma and DLBCL) was announced June 2016.[8] It is in combination trials for various leukemias and lymphomas, such as mantle cell lymphoma (MCL)[9][10] and other lymphomas.[11]
Umbralisib was granted breakthrough therapy desgination by the U.S. Food and Drug Administration (FDA) for use in people with marginal zone lymphoma (MZL), a type of cancer with no specifically approved therapies.[12]
FDA approval was based on two single-arm cohorts of an open-label, multi-center, multi-cohort trial, UTX-TGR-205 (NCT02793583), in 69 participants with marginal zone lymphoma (MZL) who received at least one prior therapy, including an anti-CD20 containing regimen, and in 117 participants with follicular lymphoma (FL) after at least two prior systemic therapies.[2] The application for umbralisib was granted priority review for the marginal zone lymphoma (MZL) indication and orphan drug designation for the treatment of MZL and follicular lymphoma (FL).[2][13][14][15][16]
SYNTHESIS
WO 2014071125
clip
First new chemical entity discovered by Indian scientists gets USFDA approval
Rhizen has retained commercialisation rights for India while also being the manufacturing and supply partner for Umbralisib. Alembic owns 50 per cent stake in Rhizen
Umbralisib, a novel cancer drug discovered and out-licensed by India’s Alembic Pharmaceuticals and its associate drug discovery company Rhizen Pharmaceuticals, has received the drug regulatory approval for sales in the US market. The drug is touted to be the first new chemical entity (NCE) discovered by Indian scientists to secure a US Food and Drug Administration (FDA) approval.
Switzerland based Rhizen had discovered the molecule in 2012 and two years later was licensed to US based TG Therapeutics, which has worldwide sales rights. Rhizen has retained commercialisation rights for India while also being the manufacturing and supply partner for Umbralisib. Alembic owns 50 per cent stake in Rhizen.
Umbralisib is a novel, next generation, oral, once daily drug for adult patients with relapsed or refractory lymphoma and relapsed or refractory marginal zone lymphoma (MZL) that resists treatments and drugs. Such cancers affect over 3-4 lakh patients in the US every year. The drug is estimated to have a global market worth US$ 1-1.5 billion.
“We are extremely proud of this historic milestone for Rhizen, and of the fact that Umbralisib is the first NCE discovered by Indian scientists to secure a US FDA approval,” said Pranav Amin, Chairman, Rhizen Pharmaceuticals & Managing Director of Alembic Pharmaceuticals.
“We are keen to bring Umbralisib to Indian patients and we plan to initiate activities towards registration and approval there soon,” said Swaroop Vakkalanka, President & CEO of Rhizen Pharmaceuticals.
Ahmedabad-based Zydus Cadila had a few months ago got ‘Fast Track Designation’ by the US Food and Drug Administration (USFDA) for Saroglitazar in the treatment of patients with Primary Biliary Cholangitis (PBC), a liver disorder due to progressive destruction of the bile ducts.
PATENT
WO 2021009509
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021009509&tab=PCTDESCRIPTION&_cid=P10-KLABTI-88387-1
Umbralisib, having the chemical designation (S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one, is an orally available PI3K delta inhibitor. Umbralisib has the following structure:
Inhibition of PI3K delta signaling with umbralisib has demonstrated activity in several pre-clinical models and primary cells from patients with hematologic malignancies. In a Phase 2 trial, umbralisib provided effective PI3K-delta inhibition and appeared well-tolerated among patients with relapsed/refractory marginal zone lymphoma. Umbralisib is currently in Phase 3 clinical development in combination with ublituximab for patients with hematologic malignancies. Hematologic malignancies are forms of cancer that begin in the cells of blood-forming tissue, such as the bone marrow, or in the cells of the immune system. Examples of hematologic cancer are acute and chronic leukemias, lymphomas, multiple myeloma and myelodysplastic syndromes. Lymphomas can include follicular lymphoma (FL), small lymphocytic lymphoma (SLL), non-Hodgkin lymphoma (NHL), and diffuse large B-cell lymphoma (DLBCL), among others. Leukemia can include chronic lymphocytic leukemia (CLL), among others. The U.S. Food and Drug Administration (FDA) has granted orphan drug designation to umbralisib for the treatment of patients with follicular lymphoma and for the treatment of patients with nodal, extranodal, and splenic marginal zone lymphoma.
U.S. Patent No. 9,150,579 discloses umbralisib and pharmaceutically acceptable salts thereof, such as 4-methylbenzenesulfonate (also known as tosylate), sulphate, hydrochloride, benzenesulfonate, maleate, and camphor sulfonate salts. U.S. Patent Nos. 9,969,740 and 10,414,773 and U.S. Patent Application Publication No. 2019/0382411 disclose solid state forms of a p-toluenesulfonic acid salt (PTSA) of umbralisib. None of these references disclose an amorphous form of umbralisib monotosylate.
An amorphous form of a compound is considered to be a solid state form that lacks long-range order relative to crystalline solid state forms of the compound. The amorphous form is chemically identical to other crystalline solid state forms but can exhibit different physical properties such as intrinsic solubility, rate of dissolution, density, mechanical property, chemical and physical stability, hygroscopicity, and morphology. The differences in intrinsic solubility also may lead to a difference in the rate of absorption, thus impacting bioavailability. Generally, amorphous compounds have a higher solubility than crystalline compounds.
EXAMPLES
Examples 1-3, which follow herein, provide embodiments of the preparation of amorphous umbralisib monotosylate.
Example 1
Preparation of Amorphous Umbralisib Monotosylate by Dry Grinding of Crystalline Umbralisib Tosylate Salt
Form I of umbralisib tosylate salt is dried under vacuum at about 40 °C in an oven for at least about 3 days to remove any residual ethyl acetate. About 30 mg of the dried umbralisib tosylate salt is ground manually using a mortar (about 6 cm in diameter) and pestle for about 3 minutes. The ground umbralisib tosylate salt is identified as being amorphous by XRPD. FIG. 1 is a representative XPRD pattern for amorphous umbralisib monotosylate prepared according to Example 1.
The amorphous umbralisib monotosylate prepared according to Example 1 is characterized by a Tg of about 51 °C, as depicted in the mDSC thermogram contained in FIG. 2.
A DVS of amorphous umbralisib monotosylate prepared according to Example 1 indicates the sample is hygroscopic, with about a 4% weight change between about 0-90% relative humidity, as depicted in FIG. 3, and less than about a 1% weight change in the sample over three cycles, as depicted in FIG. 4.
An XRPD pattern of the sample after DVS indicates that the sample is still amorphous, as depicted in FIG. 5.
Example 2
Preparation of Amorphous Umbralisib Monotosylate by Dissolution of
Crystalline Umbralisib Tosylate Salt in Methanol and Its Evaporation Therefrom
About 470 mg of Form I of umbralisib tosylate salt is dissolved in about 20 mL of methanol at about 50 °C. A solid umbralisib tosylate salt is obtained by evaporation of the solution under vacuum at about 40 °C in an oven overnight. The isolated product is identified as being amorphous umbralisib monotosylate by XRPD. FIG. 6 is a representative XPRD pattern for amorphous umbralisib monotosylate prepared according to Example 2.
The amorphous umbralisib monotosylate prepared according to Example 2 is characterized by a Tg of about 75 °C, as depicted in the mDSC thermogram contained in FIG. 7.
A TGA of amorphous umbralisib monotosylate prepared according to Example 2 shows about a 0.9% weight loss up to about 120 °C, as depicted in FIG. 8.
A DVS of amorphous umbralisib monotosylate prepared according to Example 2 indicates that the sample is hygroscopic, with about a 4% weight change between about 0-90% relative humidity, as depicted in FIG. 9, with about a 0.5% weight change in the sample over three cycles, as depicted in FIG. 10.
An XRPD pattern of the sample after DVS indicates that the sample is still amorphous, as depicted in FIG. 11.
‘ H NMR is carried out on a sample of amorphous umbralisib monotosylate prepared according to Example 2 in DMSO-d6 which indicates an umbralisib tosylate salt with a 1 :0.9 ratio of free base to acid, as depicted in FIG. 12. The peak at 8.25 ppm is representative of a single proton in the free base and the peaks at 2.30 ppm are the three protons from p-toluenesulfonic acid. A trace amount (about 0.07%) of methanol is observed at 3.16 ppm.
FTIR spectra is collected on amorphous umbralisib monotosylate prepared according to Example 2, as depicted in FIG. 13(a) and on starting crystalline umbralisib tosylate salt, as depicted in FIG. 13(b).
XRPD of amorphous umbralisib monotosylate prepared according to Example 2 after storage at about 40 °C under vacuum conditions for about two weeks indicates that the sample is still amorphous, as depicted in FIG. 14. Further, mDSC of amorphous umbralisib monotosylate after storage at about 40 °C under vacuum conditions for about two weeks indicates that the Tg is increased to about 83 °C, as depicted in FIG. 15.
Example 3
Solution Preparation of Amorphous Umbralisib Monotosylate from Umbralisib
Free Base and p-Toluenesulfonic Acid
Umbralisib free base and p-toluenesulfonic acid are each separately dissolved in MeOH. Specifically, about 72 mg of umbralisib free base is dissolved in about 3mL of MeOH at about 50 °C and about 24 mg of p-toluenesulfonic acid is dissolved in about 0.25 mL of MeOH at about 50 °C. The two solutions are mixed and stirred at room temperature for about 1 hr and then at about 4 °C overnight. The solution is transferred to a vacuum oven at about 40 °C overnight to evaporate the MeOH. Amorphous umbralisib monotosylate, identified by XRPD, is obtained. FIG. 16 is a representative XPRD pattern for amorphous umbralisib monotosylate prepared according to Example 3.
PATENT
WO 2015181728
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015181728&_cid=P10-KLABVD-88799-1
TGR-1202, chemically known as (S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one, has the following chemical structure:
[04] The preparation of TGR-1202 and its salts is described in International Publication No. WO 2014/006572 and U.S. Patent Publication No. 2014/0011819, each of which is incorporated herein by reference in its entirety for all purposes. TGR-1202 is an investigational drug currently undergoing multiple clinical trials in the area of haematological malignancies.
[05] WO 2014/006572 and US 2014/0011819 describe the synthesis of TGR-1202 (Example B l) and also disclose the therapeutic activity of this molecule to inhibit, regulate and/or modulate the signal transduction of PI3K.
Example 1: Preparation of the PTSA Salt of TGR-1202 (Form A)
[103] 7100 g of TGR-1202 was charged in a reactor containing 56.8 litres of acetone and stirred at ambient temperature. 4680 g of p-toluene sulphonic acid was added and the reaction mixture was heated at a temperature of 60-65° C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 142 litres of diethyl ether was then added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass. The solid mass was re-suspended in diethyl ether, stirred for 6 hours, and then filtered to yield a solid mass which was subsequently dissolved in 56.8 litres of acetone, filtered through a HiFlow bed, and concentrated under reduced pressure. The resulting residue mass was stirred with water overnight, then filtered and vacuum dried to yield 6600 g of the PTSA salt of TGR-1202. HPLC: 99.21% and chiral purity of 99.64:0.36 (S:R).
Example 2: Preparation of the PTSA Salt of TGR-1202 (Form B)
1000 g of TGR-1202 was charged in a reactor containing 8 litres of acetone and stirred at ambient temperature. 666 g of p-toluene sulphonic acid was then added and the reaction mixture was heated at a temperature of 60-65 °C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 20 litres of diethyl ether was added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass which was then vacuum dried to yield 1150 g of the PTSA salt of TGR-1202. HPLC: 99.33% and chiral purity: 99.61:0.39 (S:R).
PATENT
WO 2014006572
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014006572&_cid=P10-KLABZ1-89676-1
Intermediate 1
[104] Intermediate 1: 6-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one: To a solution of 2-(l-bromoethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (15.0 g,
40.84 mmol) in DMSO (150 ml), n-butanol (7.5 ml) was added and heated to 120°C for 3h. The reaction mixture was cooled to RT, quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (7.90 g, 64%). H-NMR (δ ppm, CDC13, 400 MHz): 7.85 (dd, J = 8.1, 3 Hz, 1H), 7.54 (dd, J = 9.2, 4.2 Hz, 1H), 7.47-7.37 (m, 2H), 7.15-6.98 (m, 3H), 4.74 (quintet, J = 6.8 Hz, 1H), 2.23 (d, J = 7.4 Hz, 1H), 1.54 (d, J = 6.6 Hz, 3H).
Intermediate 2
[105] Intermediate 2: 2-acetyl-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one: DMSO (5.60 ml, 79.14 mmol) was added to dichloromethane (40 ml) cooled to -78°C, followed by oxalyl chloride (3.40 ml, 39.57 mmol). After 10 min. intermediate 1 (6.00 g, 19.78 mmol) in dichloromethane (54 ml) was added dropwise and stirred for 20 min. Triethylamine (12 ml) was added and stirred for lh. The reaction mixture was quenched with water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (4.2 g, 71%) which was used as such in the next step.
Intermediate 3
OH
[106] Intermediate 3: (S)-6-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one: To intermediate 2 (2.00 g, 6.66 mmol), R-Alpine borane (0.5M in THF, 20 ml) was added and heated to 60°C for 20h. The reaction mixture quenched with aq. 2N HC1, and
extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.51 g, 75%). Enantiomeric excess: 94.2%, enriched in the fast eluting isomer (retention time: 8.78 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 4
[107] Intermediate 4: (R)-l-(6-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl 4-chlorobenzoate: To a solution of intermediate 3 (1.45 g, 4.78 mmol) in THF (15 ml), 4-chlorobenzoic acid (0.748 g, 4.78 mmol) and triphenylphosphine (1.88 g, 7.17 mmol) were added and heated to 45 C followed by diisopropylazodicarboxylate (1.4ml, 7.17 mmol). After lh, the reaction mixture was concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.81 g, 86%) which was used without purification in the next step.
Intermediate 5
Method A
[108] Intermediate 5: (R)-6-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one: To intermediate 4 (1.75 g, 3.96 mmol) in methanol (17 ml) cooled to 10°C, potassium carbonate (0.273 g, 1.98 mmol) was added and stirred for 30 min. The reaction mixture was concentrated, acidified with 2N HC1 solution, extracted with ethyl acetate, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (1.05 g, 87%). Enantiomeric excess: 93.6%, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Method B:
[109] Step-1 : (R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one : To l-(5-fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone (11.00 g, 44.31 mmol ) in dichloromethane, HATU (33.7 g, 88.63 mmol) and R-(+)2-benzyloxypropionic acid (9.58 g, 53.17 mmol) were added and stirred for 10 min. Triethylamine (66.7 ml, 0.47 mol) was added dropwise and stirred at RT for 24h. The reaction mixture was quenched with water, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (10.5 g, 60%). ‘H-NMR (δ ppm, CDC13, 400 MHz): 7.85 (dd, J = 8.1,3 Hz, 1H), 7.58 (dd, J = 9.1, 4.1 Hz, 1H), 7.47-7.39 (m, 1H), 7.39-7.34 (m, 1H), 7.28-7.20 (m, 3H), 7.20-7.14 (m, 2H), 7.16-7.07 (m, 1H), 6.99-6.89 (m, 2H), 4.50-4.31 (m, 3H), 1.56 (d, J = 6.4 Hz, 3H).
[110] Step-2 : (R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (10.5 g, 26.69 mmol) in dichloromethane (110 ml) cooled to 0°C, aluminium chloride (5.35 g, 40.03 mmol) was added portionwise and stirred at RT for 6h. The reaction mixture was quenched with 2N HC1 solution, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the desired intermediate as a yellow solid (6.1 g, 76%). Enantiomeric excess: 97.7%, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 13
[121] Intermediate 13: 3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-amine: To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF 110 ml), ethanol (55 ml) and water (55 ml), intermediate 12 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min. Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h, the reaction mixture was filtered though celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
Example Bl
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
[127] To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate ( 0.15 ml, 0.749 mmol) was added heated to 45°C. After 2h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 %). MP: 139-142°C. Mass : 571.7 (M H-NMR (δ ppm, CDC13, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J = 8.2,3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J = 8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.11 (q, J = 7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J = 6.1 Hz, 1H), 2.00 (d, J = 7.1Hz, 3H), 1.42 (d, J = 6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC
on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64min.).
PATENT
US 2014/0011819 describe the synthesis of TGR-1202 (Example B l)
PATENT
US 20150290317
US 20150174263
WO 2014071125
WO 2014006572
WO 2013188763*
^Clinical trial number NCT02268851 for “A Phase I/Ib Safety and Efficacy Study of the PI3K-delta Inhibitor TGR-1202 and Ibrutinib in Patients With CLL or MCL” at ClinicalTrials.gov
^Clinical trial number NCT02793583 for “Study to Assess the Efficacy and Safety of Ublituximab + TGR-1202 With or Without Bendamustine and TGR-1202 Alone in Patients With Previously Treated Non-Hodgkin’s Lymphoma (UNITY-NHL)” at ClinicalTrials.gov
Feb. 9, 2021 04:45 UTC Rhizen Pharmaceuticals AG Announces That Its Partnered Asset, Umbralisib (UKONIQ™), Has Received US FDA Accelerated Approval for Adult Patients With Relapsed or Refractory MZL & FL
Umbralisib (UKONIQ™) granted accelerated approval by US FDA for the treatment of adult patients with relapsed or refractory marginal zone lymphoma (MZL), follicular lymphoma (FL).
Umbralisib, a novel next generation inhibitor of PI3K delta & CK1 epsilon, was discovered by Rhizen Pharmaceuticals and subsequently licensed to TG Therapeutics, who led the asset’s clinical development.
Rhizen and its affiliate Alembic Pharma to support TG Therapeutics towards UKONIQ’s commercialization as its manufacturing & supply partner; Rhizen plans to register and commercialize Umbralisib in India.
BASEL, Switzerland–(BUSINESS WIRE)–Rhizen Pharmaceuticals, a clinical-stage oncology-focused biopharmaceutical company, today announced that its novel next generation PI3K-delta inhibitor, Umbralisib, which was licensed to TG Therapeutics (NASDAQ:TGTX), has secured US FDA accelerated approval for the treatment of:
adult patients with relapsed or refractory marginal zone lymphoma (MZL) who have received at least one prior anti-CD20 based regimen, and
adult patients with relapsed or refractory follicular lymphoma (FL) who have received at least three prior lines of systemic therapy.
Accelerated approval was granted for these indications, under a priority review (MZL), based on the results of the Phase 2 UNITY-NHL Trial (NCT02793583); in MZL, an ORR of 49% with 16% complete responses and in FL an ORR of 43% with 3% complete responses were achieved, respectively. Umbralisib was earlier granted Breakthrough Therapy Designation (BTD) for the treatment of MZL and orphan drug designation (ODD) for the treatment of MZL and FL.
Umbralisib is a novel, next generation, oral, once daily, inhibitor of phosphoinositide 3 kinase (PI3K) delta and casein kinase 1 (CK1) epsilon and was discovered by Rhizen Pharma and subsequently licensed to TG Therapeutics (NASDAQ:TGTX) at an IND stage (TGR 1202) in 2012. In 2014, both parties entered into a licensing agreement as a part of which TGTX obtained worldwide rights and Rhizen has retained commercialization rights for India while also being the manufacturing and supply partner for Umbralisib.
Swaroop Vakkalanka, President & CEO of Rhizen Pharmaceuticals said: “Umbralisib’s approval offers MZL & FL patients a new treatment option and is a huge validation of Rhizen’s drug discovery & development capabilities. This is a momentous occasion in Rhizen’s journey as a successful biotech that speaks of the true ability of our team to discover & develop safe and effective therapies that can last the rigors of drug development. Further, we are keen to bring Umbralisib to Indian patients and we plan to initiate activities towards registration and approval there soon.”
Pranav Amin, Chairman, Rhizen Pharmaceuticals & Managing Director of Alembic Pharmaceuticals Ltd said: “We are extremely proud of this historic milestone for Rhizen, and of the fact that Umbralisib is the first NCE discovered by Indian scientists to secure a US FDA approval. We are committed to working together with TG Therapeutics and Rhizen Pharma to ensure uninterrupted supply of UKONIQ™. Umbralisib is the first discovery asset to come out of Rhizen’s R&D efforts and this approval heralds the promise of the rest of Rhizen’s deep pipeline and continuing efforts.”
About Umbralisib:
Umbralisib is the first and only oral inhibitor of phosphoinositide 3 kinase (PI3K) delta and casein kinase 1 (CK1) epsilon. PI3K-delta is known to play an important role in supporting cell proliferation and survival, cell differentiation, intercellular trafficking and immunity and is expressed in both normal and malignant B-cells. CK1-epsilon is a regulator of oncoprotein translation and has been implicated in the pathogenesis of cancer cells, including lymphoid malignancies. Umbralisib is indicated for the treatment of adult patients with relapsed or refractory marginal zone lymphoma (MZL) who have received at least one prior anti-CD20-based regimen and for the treatment of adult patients with relapsed or refractory follicular lymphoma (FL) who have received at least three prior lines of systemic therapy. These indications are approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. More information on Umbralisib or UKONIQ™ can be found at https://www.tgtherapeutics.com/prescribing-information/uspi-ukon.pdf.
About Alembic Pharmaceuticals Ltd:
Alembic Pharmaceuticals Limited, a vertically integrated research and development pharmaceutical company, has been at the forefront of healthcare since 1907. Headquartered in India, Alembic is a publicly listed company that manufactures and markets generic pharmaceutical products all over the world. Alembic’s state of the art research and manufacturing facilities are approved by regulatory authorities of many developed countries including the USFDA. Alembic is one of the leaders in branded generics in India. Alembic’s products that are marketed through a marketing team of over 5000 are well recognized by doctors and patients.
Information about Alembic can be found at http://www.alembicpharmaceuticals.com/.
(Reuters: ALEM.NS) (Bloomberg: ALPM) (NSE: APLL TD) (BSE: 533573)
About Rhizen Pharmaceuticals A.G.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel onco-therapeutics. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways. Rhizen is headquartered in Basel, Switzerland. For additional information, please visit www.rhizen.com.
View source version on businesswire.com: https://www.businesswire.com/news/home/20210208005742/en/ Contacts
////////////ウムブラリシブトシル酸塩 , Umbralisib, fda 2021, 2021 approvals, TGR 1202, TGR-1202-101, RP 5264, Umbralisib tosylate, RP-5307 , TGR-1202, TGR-1202 PTSA, FU8XW5V3FS , RP-5264, AK173784,
old post pasted
A PI3K inhibitor potentially for treatment of chronic lymphocytic leukemia, leukemia,lymphoma,B-cell
TGR‐1202, a next generation PI3K-δ delta inhibitor. TGR-1202 (RP-5264) is a highly specific, orally available, PI3K delta inhibitor, targeting the delta isoform with nanomolar potency and several fold selectivity over the alpha, beta, and gamma isoforms of PI3K.
TG Therapeutics, under license from Rhizen Pharmaceuticals, is developing TGR-1202 (structure shown; formerly RP-5264), a lead from a program of PI3K delta inhibitors, for the potential oral treatment of hematological cancers including Hodgkin lymphoma, non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), B-cell lymphoma and mantle cell lymphoma (MCL)
Incozen Therapeutics Pvt Ltd
TG Therapeutics
TGR-1202 potential to perform as the best PI3K inhibitor in its class and the possible superiority of TG-1101 over Rituxan®.
Rhizen Pharmaceuticals S.A.
Description
Phosphoinositide 3-kinase (PI3K) delta inhibitor
Leukemia, chronic lymphocytic PHASE 3, TG Therapeutics
Orphan Drug
Umbralisib is a novel phosphatidylinositol 3-kinase delta (PI3Kdelta) inhibitor under development at TG Therapeutics in phase III clinical trials, in combination with ublituximab, for the treatment of chronic lymphocytic leukemia (CLL) and for the treatment of diffuse large B-cell lymphoma (DLBCL). The company refers to the combination regimen of ublituximab and TGR-1202 as TG-1303. The drug is also in phase II clinical development for the oral treatment of hematologic malignancies, as a single agent or in combination therapy. Phase I clinical trials are ongoing in patients with select relapsed or refractory solid tumors, such as adenocarcinoma of the pancreas, adenocarcinoma of the colon, rectum, gastric and GE junction cancer, and GI Stromal Tumor (GIST).
In 2016, orphan drug designation was assigned to the compound in the U.S. for the treatment of CLL. In 2017, additional orphan drug designation was granted in the U.S. for the treatment of CLL and DLBCL, in combination with ublituximab.
Originated by Rhizen Pharmaceuticals, the product was jointly developed by Rhizen Pharmaceuticals and TG Therapeutics since 2012. In 2014, exclusive global development and commercialization rights (excluding India) were licensed to TG Therapeutics.
Rhizen Pharmaceuticals Announces Out-licensing Agreement for TGR-1202, a Novel Next Generation PI3K-delta Inhibitor
Rhizen to receive upfront payment of $8.0 million — Rhizen to retain global manufacturing and supply rights — Rhizen to retain development and commercialization for India
Rhizen to retain development and commercialization for India
September 23, 2014 09:00 ET | Source: Rhizen Pharmaceuticals SA
La Chaux-de-Fonds, Switzerland, Sept. 23, 2014 (GLOBE NEWSWIRE) — Rhizen Pharmaceuticals S.A. today announced an out-licensing agreement for TGR-1202, a novel next generation PI3K-delta inhibitor. TG Therapeutics exercised its option for early conversion to a licensing agreement from a 50:50 joint venture partnership.
In exchange for this licensing agreement, TG Therapeutics will pay Rhizen an upfront payment of $8.0 million ($4.0 million in cash and $4.0 million in TG Therapeutics common stock). In addition to the upfront payment, Rhizen will be eligible to receive regulatory filing, approval and sales based milestones in the aggregate of approximately $240 million, and tiered royalties based on net sales.
Swaroop Vakkalanka, Ph.D. and President of Rhizen stated, “We are extremely happy and take pride in discovering a novel, next generation, once-daily PI3K-delta inhibitor under active development led by TG Therapeutics. We are encouraged by the progress of TRG-1202 to date, and the speed at which TG Therapeutics is developing the asset in various hematological malignancies. We look forward to the day this novel drug reaches cancer patients in need of new and safe therapies.”
About Rhizen Pharmaceuticals S.A.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel therapeutics for the treatment of cancer, immune and metabolic disorders. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways. Rhizen is headquartered in La-Chaux-de-Fonds, Switzerland. For additional information, please visit Rhizen’s website, www.rhizen.com.
TGR-1202.with Idelalisib and IPI-145 (left to right) for comparison.
IPI 145
PATENT
WO 2015181728
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015181728
TGR-1202, chemically known as (S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one, has the following chemical structure:
Example 1: Preparation of the PTSA Salt of TGR-1202 (Form A)
7100 g of TGR-1202 was charged in a reactor containing 56.8 litres of acetone and stirred at ambient temperature. 4680 g of p-toluene sulphonic acid was added and the reaction mixture was heated at a temperature of 60-65° C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 142 litres of diethyl ether was then added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass. The solid mass was re-suspended in diethyl ether, stirred for 6 hours, and then filtered to yield a solid mass which was subsequently dissolved in 56.8 litres of acetone, filtered through a HiFlow bed, and concentrated under reduced pressure. The resulting residue mass was stirred with water overnight, then filtered and vacuum dried to yield 6600 g of the PTSA salt of TGR-1202. HPLC: 99.21% and chiral purity of 99.64:0.36 (S:R).
Example 2: Preparation of the PTSA Salt of TGR-1202 (Form B)
1000 g of TGR-1202 was charged in a reactor containing 8 litres of acetone and stirred at ambient temperature. 666 g of p-toluene sulphonic acid was then added and the reaction mixture was heated at a temperature of 60-65 °C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 20 litres of diethyl ether was added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass which was then vacuum dried to yield 1150 g of the PTSA salt of TGR-1202. HPLC: 99.33% and chiral purity: 99.61:0.39 (S:R).
Table 1 lists the XRPD pattern peaks and relative peak intensities for the products of Examples 1 and 2.
TABLE 1
The tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 2 exhibited a Cmax about 2.5 fold and an area under the curve (AUC) about 1.9 fold greater than that of the tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 1. The results are provided in Table 8 below.
TABLE 8
PATENT
WO 2014071125
http://www.google.com/patents/WO2014071125A1?cl=en
formula (A) that is a ΡΒΚδ selective inhibitor,
(A)
Synthesis of Compound of Formula A
Unless otherwise stated, purification implies column chromatography using silica gel as the stationary phase and a mixture of petroleum ether (boiling at 60-80°C) and ethyl acetate or dichloromethane and methanol of suitable polarity as the mobile phases. The term “RT” refers to ambient temperature (25-28°C).
Intermediate 1 : 2-( l-bromoethyl)-6-fluoro-3-f3-fluorophenyl)-4H-chromen-4-one
Step-1 [l-(5-Fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone]: 3- Fluorophenylacetic acid (7.33 g, 47.56 mmoles) was dissolved in 25 ml dichloromethane. To this mixture, oxalylchloride (7.54 g, 59.46 mmoles) and DMF (3 drops) were added at 0°C and stirred for 30 min. The solvent was evaporated and dissolved in 25 ml dichloromethane. To this mixture, 4-fluoroanisole (5.00 g, 39.64 mmoles) was added and cooled to 0°C. At 0°C A1C13 (7.95 g, 59.46 mmoles) was added and the reaction mixture was warmed to RT and stirred for 12 hours. The reaction mixture was quenched by the addition of 2N HC1, extracted with ethyl acetate, dried over sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as colorless solid (4.5 g, 45% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 11.34 (s, 1H), 7.75 (dd, J=9.4, 3.1 Hz, 1H), 7.42 (m, 2H), 7.12 (m, 3H), 7.05 (dd, J=9.0, 4.5 Hz, 1H), 4.47 (s, 2H).
Step-2 [2-Ethyl-6-fiuoro-3-(3-fluorophenyl)-4H-chromen-4-one]: l-(5-Fluoro-2- hydroxyphenyl)-2-(3-fluorophenyl)ethanone obtained from Step-1 (3.00 g, 12.08 mmoles) was placed in a round bottom flask and to this triethylamine (25 ml) and propionic anhydride (4.92 g, 37.82 mmoles) were added, and the mixture was refluxed for 24 hours. After cooling to RT, the reaction mixture was acidified by the addition of IN HC1 solution, extracted with ethyl acetate, washed with sodium bicarbonate solution, dried with sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as off-yellow solid (1.80 g, 52% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.80 (m, 1H), 7.76 (m, 2H), 7.51 (dd, J=8.0, 6.4 Hz), 7.22 (m, 1H), 7.18 (m, 2H), 2.56 (q, J=7.6 Hz, 2H), 1.20 (t, J=7.6 Hz, 3H).
Step-3: To a solution of 2-Ethyl-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained from Step-2 (1.80 g, 6.28 mmoles) in carbon tetrachloride (20 ml), N- bromosuccinimide (1.11 g, 6.28 mmoles) was added and heated to 80°C. Azobisisobutyronitrile (10 mg) was added to the reaction mixture at 80°C. After 12 hours, the reaction mixture was cooled to RT, diluted with dichloromethane and washed with water. The organic layer was dried over sodium sulphate and concentrated under reduced pressure to afford the crude title compound as yellow solid (1.25 g, 55% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.91 (dd, J=9.2, 4.3 Hz, 1H), 7.81 (dt, j=8.2, 2.8 Hz, 1H), 7.74 (dd, J=8.3, 3.1 Hz, 1H), 7.57 (m, 1H), 7.32 (dt, J=8.5, 2.4 Hz, 1H), 7.19 (m, 2H), 5.00 (q, J=6.8 Hz, 1H), 1.97 (d, J=6.8 Hz, 3H).
Intermediate 2: 6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one
To a solution of Intermediate 1 (15.0 g, 40.84 mmol) in DMSO (150 ml), n-butanol (7.5 ml) was added and heated to 120°C for 3 hours. The reaction mixture was cooled to RT, quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (7.90 g, 64%). 1H-NMR (δ ppm, CDC13, 400 MHz): 7.85 (dd, J = 8.1, 3 Hz, 1H), 7.54 (dd, J = 9.2, 4.2 Hz, 1H), 7.47-7.37 (m, 2H), 7.15-6.98 (m, 3H), 4.74 (quintet, J= 6.8 Hz, 1H), 2.23 (d, J = 7.4 Hz, 1H), 1.54 (d, J = 6.6 Hz, 3H).
Intermediate 3 : 2-acetyl-6-fluoro-3-( 3-fluorophenyl)-4H-chromen-4-one
DMSO (5.60 ml, 79.14 mmol) was added to dichloromethane (40 ml), and cooled to – 78°C, followed by oxalyl chloride (3.40 ml, 39.57 mmol). After 10 min., intermediate 2 (6.00 g, 19.78 mmol) in dichloromethane (54 ml) was added dropwise and stirred for 20 min.
Triethylamine (12 ml) was added and stirred for 1 hour. The reaction mixture was quenched with water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (4.2 g, 71%) which was used as such in the next step.
Intermediate 4: fS)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one
To intermediate 3 (2.00 g, 6.66 mmol), R-Alpine borane (0.5 M in THF, 20 ml) was added and heated to 60°C for 20 hours. The reaction mixture quenched with 2N HC1, and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.51 g, 75%).
Enantiomeric excess: 94.2%, enriched in the fast eluting isomer (retention time: 8.78 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 5: fR)-l-f6-fluoro-3-f3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl 4- chlorobenzoate
To a solution of intermediate 4 (1.45 g, 4.78 mmol) in THF (15 ml), 4-chlorobenzoic acid (0.748 g, 4.78 mmol) and triphenylphosphine (1.88 g, 7.17 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (1.4 ml, 7.17 mmol). After 1 hour, the reaction mixture was concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.81 g, 86%) which was used without purification in the next step. Intermediate 6: fR)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one
Method A
Intermediate 5 (1.75 g, 3.96 mmol) in methanol (17 ml) was cooled to 10°C, potassium carbonate (0.273 g, 1.98 mmol) was added and stirred for 30 min. The reaction mixture was concentrated, acidified with 2N HCl solution, extracted with ethyl acetate, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (1.05 g, 87% yield). Enantiomeric excess: 93.6%>, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Method B
Step-1 [(R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one]: To l-(5-fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone (11.00 g, 44.31 mmol) in dichloromethane, HATU (33.7 g, 88.63 mmol) and R-(+)2-benzyloxypropionic acid (9.58 g, 53.17 mmol) were added and stirred for 10 min. Triethylamine (66.7 ml, 0.47 mol) was added dropwise and stirred at RT for 24 hours. The reaction mixture was quenched with water, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:
petroleum ether to afford the title compound as a yellow solid (10.5 g, 60%> yield). 1H-NMR (δ ppm, CDCls, 400 MHz): 7.85 (dd, J = 8.1,3 Hz, 1H), 7.58 (dd, J = 9.1, 4.1 Hz, 1H), 7.47-7.39 (m, 1H), 7.39-7.34 (m, 1H), 7.28-7.20 (m, 3H), 7.20-7.14 (m, 2H), 7.16-7.07 (m, 1H), 6.99-6.89 (m, 2H), 4.50-4.31 (m, 3H), 1.56 (d, J = 6.4 Hz, 3H).
Step-2: (R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained in Step-1 (10.5 g, 26.69 mmol) in dichloromethane (110 ml) was cooled to 0°C, aluminium chloride (5.35 g, 40.03 mmol) was added portionwise and stirred at RT for 6 hours. The reaction mixture was quenched with 2N HCl solution, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford intermediate 6 a yellow solid (6.1 g, 76% yield). Enantiomeric excess: 97.7%, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 7: 4-bromo-2-fluoro-l-isopropoxybenzene
To a solution of 4-bromo-3-fluorophenol (10 g, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8 ml, 62.62 mmol) and triphenylphosphine (20.6 g, 78.52 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (15.4 ml, 78.52 mmol). The mixture was refluxed for 1 hour, concentrated and the residue was purified by column
chromatography with ethyl acetate: petroleum ether to afford the title compound as a colorless liquid (13.1 g, 99% yield), which was used without purification in the next step.
Intermediate 8: 2-f3-fluoro-4-isopropoxyphenyl)-4,4,5.,5-tetramethyl-l,3i2-dioxaborolane
Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15 g, 58.96 mmol) were added to a solution of intermediate 7 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [l, -Bis(diphenylphosphino)ferrocene]dichloro palladium(II) CH2CI2 (4.4 g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 9: 3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-dlpyrimidin-4-amine
To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF (110 ml), ethanol (55 ml) and water (55 ml), intermediate 8 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min.
Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
(RS)- 2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
To a solution of intermediate 9 (0.080 g, 0.293 mmol) in DMF (2 ml), potassium carbonate (0.081 g, 0.587 mmol) was added and stirred at RT for 10 min. To this mixture intermediate 1 (0.215 g, 0.587 mmol) was added and stirred for 12 hours. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a pale yellow solid (0.045 g). MP: 175-177°C. 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 8.20 (s, 1H), 7.85 (dd, J = 81, 3.0 Hz, 1H), 7.48-7.33 (m, 5H), 7.14 (t, J= 8.3 Hz, 1H), 7.02 (m, 2H), 6.90 (m, 1H), 6.10 (q, J = 7.1 Hz, 1H), 5.42 (s, 2H), 4.64 (quintet, J = 6.0 Hz, 1H), 1.99 (d, J = 7.1 Hz, 3H), 1.42 (d, J= 6.1 Hz, 6H).
fS)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (“S-isomer”)
To a solution of intermediate 9 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 6 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45°C. After 2 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 % yield). MP: 139-142°C. Mass: 571.7 (M+). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64 min.). fR)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-ehromen-4-one
To a solution of intermediate 8 (0.284 g, 0.989 mmol) in THF (5.0 ml), intermediate 4 (0.250 g, 0.824 mmol) and tris(4-methoxy)phenylphosphine (0.435 g, 1.23 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.25 ml, 1.23 mmol) was added stirred at RT. After 12 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate :
petroleum ether to afford the title compound as an off-white solid (0.105 g, 22 % yield). MP: 145-148°C. Mass: 571.7 (M+). Enantiomeric excess: 95.4% as determined by HPLC on a chiralpak AD-H column, enriched in the late eluting isomer (retention time = 14.83 min.).
PATENT
WO 2014006572
http://www.google.com/patents/WO2014006572A1?cl=enB1 IS DESIRED
(S)-2- (l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-6- fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (compound-B l)
Intermediate 11
[119] Intermediate 11: 4-bromo-2-fluoro-l-isopropoxybenzene:To a solution of 4-bromo-2- fluorophenol (lOg, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8ml, 62.62 mmol) and triphenylphosphine (20.6g, 78.52 mmol) were added and heated to 45 C followed by diisopropylazodicarboxylate (15.4ml, 78 52 mmol). The mixture was refluxed for lh, concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a colourless liquid (13. lg, 99%) which was used without purification in the next step. Intermediate 12
[120] Intermediate 12: 2-(3-fluoro-4-isopropoxyphenyl)-4,4,5,5-tetramethyl- 1,3,2- dioxaborolane: Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15g, 58.96 mmol) were added to a solution of intermediate 11 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [1,1 ‘- Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH2Cl2 (4.4g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 13
[121] Intermediate 13: 3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- amine: To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF 110 ml), ethanol (55 ml) and water (55 ml), intermediate 12 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min. Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h, the reaction mixture was filtered though celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
Example Bl
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
[127] To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate ( 0.15 ml, 0.749 mmol) was added heated to 45°C. After 2h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 %). MP: 139- 142°C. Mass : 571.7 (M H-NMR (δ ppm, CDC13, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J = 8.2,3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J = 8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.11 (q, J = 7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J = 6.1 Hz, 1H), 2.00 (d, J = 7.1Hz, 3H), 1.42 (d, J = 6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64min.).
PATENT
US 2014/0011819 describe the synthesis of TGR-1202 (Example B l) http://www.google.co.in/patents/US20140011819
Example B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45° C. After 2 h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:petroleum ether to afford the title compound as an off-white solid (0.049 g, 20%). MP: 139-142° C. Mass: 571.7 (M+).1H-NMR (δ ppm, CDCl3, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J=8.2, 3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J=8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J=8.4 Hz, 1H), 6.11 (q, J=7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J=6.1 Hz, 1H), 2.00 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=10.64 min)
4-Methylbenzenesulfonate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (22.7 g, 39.69 mmol) in isopropanol (600 ml), p-toluenesulphonic acid (8.30 g, 43.66 mmol) was added and refluxed for 1 h. The reaction mixture was concentrated, co-distilled with petroleum ether and dried. To the residue water (300 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (28.2 g, 95%). MP: 138-141° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.80 (d, J=8.2 Hz, 2H), 7.51 (dd, J=9.3, 4.3 Hz, 1H), 7.45 (dd, J=7.5, 3.1 Hz, 1H), 7.42-7.31 (m, 3H), 7.29 (m, 2H), 7.22 (d, J=8.0 Hz, 2H), 7.16 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.5 Hz, 1H), 6.97 (br s, 1H), 6.88 (br s, 1H), 6.11 (q, J=7.2 Hz, 1H), 4.67 (quintet, J=6.0 Hz, 1H), 2.36 (s, 3H), 2.03 (d, J=7.1 Hz, 3H), 1.43 (d, J=6.0 Hz, 6H). Mass: 572.4 (M++1-PTSA). Enantiomeric excess: 93.4% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.35 min.)
Sulphate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulfate
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulphate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (15.0 g, 26.24 mmol) in isopropanol (600 ml) was cooled to 0° C. To this Sulphuric acid (2.83 g, 28.86 mmol) was added and stirred at room temperature for 24 h. The reaction mass was filtered and washed with petroleum ether and dried under vacuum. To the solid, water (150 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (13.5 g, 76%). MP: 125-127° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.51 (dd, J=9.2, 4.2 Hz, 1H), 7.45-7.31 (m, 3H), 7.29 (m, 1H), 7.15 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.4 Hz, 1H), 6.96 (br s, 1H), 6.88 (br s, 1H), 6.09 (q, J=7.1 Hz, 1H), 4.676 (quintet, J=6.1 Hz, 1H), 2.01 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Mass: 572.2 (M++1-H2SO4). Enantiomeric excess: 89.6% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.08 min.)
Various other acid addition salts of compound B1 were prepared as provided in Table 1.
Rhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) …
LFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer Molecular target: CD20 Description: Glycoengineered mAb against CD20 …
TG Therapeutics Inc. (NASDAQ:TGTX) rose $2.65 (23%) to $14.37 after the company said it received an SPA from FDA for the Phase III UNITY-CLL trial of ublituximab (TG-1101) in combination with TGR-1202 to treat chronic …
Now that Nimbus has put IRAK4 on the map for B cell lymphoma, several companies are closing in with their own inhibitors, and they’re all on track for IND-enabling studies this year.
Rhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) …
LFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Ildong Pharmaceutical Co. Ltd. (KSE:000230), Seoul, South Korea Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer …
Rhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) …
Rhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Business: Cancer TG Therapeutics exercised an option under a 2012 deal to license exclusive, worldwide …
Treatment Of Cancers Using PI3 Kinase Isoform Modulators [US2014377258]
2014-05-30
2014-12-25
////////Umbralisib
CC(C)OC1=C(C=C(C=C1)C2=NN(C3=C2C(=NC=N3)N)C(C)C4=C(C(=O)C5=C(O4)C=CC(=C5)F)C6=CC(=CC=C6)F)F
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021164789&_cid=P12-KSZQ3G-94695-1Phosphatidylinositol 3-kinase (phosphatidylinositol-3-kinase, PI3K) is composed of the regulatory subunit p85 or p101, and the catalytic subunit p110 (subdivided into four subtypes: p110a, p110b, p110g, and p110d) Lipid kinase catalyzes the phosphorylation of the inositol ring 3′-OH of phosphatidylinositol 4,5-bisphosphate (phosphatidylinositol 4,5-bisphosphate, PIP2) to phosphatidylinositol 3,4,5-triphosphate (phosphatidylinositol 4,5-bisphosphate, PIP2). 3,4,5-trisphosphate, PIP3) and activate downstream Akt, which plays a key role in cell proliferation, survival and metabolism. In tumor cells, PI3K is overexpressed, which leads to rapid proliferation and growth of tumor cells.
The tumor suppressor gene PTEN (phosphatase, tension homolog deleted on chromosome ten) dephosphorylates PIP3 to generate PIP2, which leads to negative feedback regulation of the PI3K signaling pathway, inhibits cell proliferation and promotes cell apoptosis. PI3K gene mutation and amplification frequently occur in cancer, and PTEN gene deletion in cancer, etc., suggest that PI3K overexpression is closely related to tumorigenesis.
TGR-1202 is a second-generation PI3Kδ inhibitor developed by TG Therapeutic. Compared with the first-generation PI3Kδ inhibitor, it can significantly reduce the toxicity of liver and gastrointestinal tract in clinical trials, and patients with large B-cell lymphoma are also exposed to TGR. -1202 There is a partial response. Patent WO2014006572 discloses the structure of TGR-1202. ACP-196 is a second-generation BTK inhibitor that has been approved for marketing by the FDA. It has been reported in the literature (PLoS ONE 12(2):e0171221.). The combination of a PI3Kδ inhibitor and a BTK inhibitor can inhibit BCR signaling in two ways. Access, thereby playing a synergistic effect.Example 1: Preparation of the compound of formula (I)
Step 1: Synthesis of compound BB-1-3
To a solution of BB-1-1 (23g, 205.17mmol, 1eq) in polyphosphoric acid (23g, 17.84mmol) was added BB-1-2 (43.90g, 266.71mmol, 1.3eq). The reaction solution was stirred at 125°C for 5 hours under the protection of nitrogen. After the completion of the reaction, water (300 mL) was added to the reaction solution to quench the reaction, and a solid precipitated out, which was directly filtered to obtain a filter cake. The filter cake was washed with water (100 mL) once, and then purified by column chromatography (PE:EA=1:1) to obtain the target compound BB-1-3. 1 H NMR (400MHz, CDCl 3 ) δ 8.94 (br s, 1H), 7.68 (br d, J=5.3 Hz, 2H), 6.65 (s, 1H), 4.51 (s, 2H).
Step 2: Synthesis of compound BB-1-4
To a solution of BB-1-3 (21.02g, 98.87mmol, 1eq) in glacial acetic acid (210mL) was added NBS (19.36g, 108.75mmol, 1.1eq). The reaction solution was stirred at 25°C for 1 hour under the protection of nitrogen. After the completion of the reaction, water (200 mL) was added to the reaction solution to quench the reaction, and a solid was formed, which was filtered to obtain a filter cake. After washing three times with water (30mL*3), the filter cake was dissolved in dichloromethane (100mL), dried over anhydrous sodium sulfate, concentrated, and then beaten with methyl tert-butyl ether (50mL) once. The filter cake was collected by filtration to obtain the target Compound BB-1-4 batch one. The aqueous phase was extracted with dichloromethane (100mL*3) and combined with the mother liquor obtained by washing with methyl tert-butyl ether, and then subjected to column chromatography (petroleum ether: ethyl acetate = 1:1, target product Rf = 0.43) ) Purification to obtain the target compound BB-1-4 batch two. The two batches were dissolved and combined with dichloromethane and spin-dried to obtain the target compound BB-1-4. 1 H NMR (400 MHz, CDCl 3 ) δ 8.93 (dd, J = 1.3, 3.1 Hz, 1H), 7.80-7.69 (m, 2H), 4.74 (s, 2H).
Step 3: Synthesis of compound BB-1-5
To a solution of BB-1-4 (3g, 10.29mmol, 1eq) in N,N-dimethylformamide (30mL), potassium acetate (1.52g, 15.44mmol, 1.5eq) was added. The reaction solution was stirred at 40°C for 3.5 hours under the protection of nitrogen. After the reaction was completed, water (60 mL) was added to the reaction solution to quench the reaction, and a large amount of solid was formed, which was filtered to obtain a filter cake. The filter cake was dissolved in dichloromethane (100 mL), dried over anhydrous sodium sulfate, and concentrated to obtain the target compound BB-1-5 batch one. The aqueous phase was extracted with methyl tert-butyl ether (100 mL*3) to obtain the organic phase, dried over anhydrous sodium sulfate, and concentrated to obtain BB-1-5 batch two, which was obtained by combining the two batches. Used directly in the next reaction. 1 H NMR (400MHz, CDCl 3 ) δ 9.07-8.88 (m, 1H), 7.71 (dd, J=1.7, 5.8 Hz, 2H), 5.31-5.26 (m, 2H), 2.22 (s, 3H).
Step 4: Synthesis of compound BB-1-6
To the dioxane (37 mL) solution of BB-1-5 (3.77 g, 11.96 mmol, 1 eq), hydrochloric acid (12M, 3.49 mL, 3.5 eq) was added. The reaction solution was stirred at 40°C for 3.5 hours under the protection of nitrogen. After the completion of the reaction, the reaction solution was concentrated, water (2 mL) was added, the pH was adjusted to 9 with ammonia water, and the filter cake was collected by filtration. After dissolving with dichloromethane (100 mL), drying with anhydrous sodium sulfate, and concentration, the target compound BB-1-6 was obtained. Used directly in the next reaction. 1 H NMR(400MHz,DMSO-d 6 )δ8.95(dd,J=2.9,4.6Hz,1H), 8.15(dd,J=2.6,7.0,9.6Hz,1H), 7.86(dd,J=5.3 , 9.6 Hz, 1H), 5.35 (t, J = 5.9 Hz, 1H), 4.58 (d, J = 6.1 Hz, 2H).
Step 5: Synthesis of compound BB-1-7
To BB-1-6 (2.6g, 9.52mmol, 1eq) and 3-fluorophenylboronic acid (2.66g, 19.04mmol, 2eq) in acetonitrile/water (12.5mL, volume ratio: 3/1), add carbonic acid Sodium ( 5.05g, 47.61mmol, 5eq ) and Pd(PPh 3 ) 4 (550.15mg, 476.09μmol, 0.05eq). The reaction solution was stirred at 85°C for 4 hours under the protection of nitrogen. After the reaction was completed, dichloromethane (50 mL) was added to the reaction solution, and then water (5 mL) was slowly added to quench the reaction, and then extracted with dichloromethane (50 mL*3). The organic phases were combined, dried over anhydrous sodium sulfate, concentrated, and purified by column chromatography (petroleum ether: ethyl acetate=0:1) to obtain BB-1-7. 1 H NMR(400MHz,DMSO-d 6 )δ8.94(dd,J=3.1,4.8Hz,1H), 8.12(ddd,J=2.6,7.1,10.0Hz,1H), 7.84(dd,J=5.3 , 10.1 Hz, 1H), 7.62 (s, 1H), 7.27-7.15 (m, 3H), 5.25 (t, J = 5.9 Hz, 1H), 4.28 (d, J = 5.7 Hz, 2H).
Step 6: Synthesis of compound BB-1-8
In a three-neck flask, at -78°C, to a solution of oxalyl chloride (1.85g, 14.57mmol, 1.28mL, 3eq) in dichloromethane (20mL) was added DMSO (2.28g, 29.14mmol, 2.28mL, 6eq). The reaction solution was stirred at -78°C for 1 hour under the protection of nitrogen. A solution of BB-1-7 (1.4g, 4.86mmol, 1eq) in dichloromethane (20mL) was added, and the mixture was stirred at -78°C for 1 hour. Triethylamine (4.91g, 48.57mmol, 6.76mL, 10eq) was added, and after stirring at -78°C for 1 hour, the reaction solution was raised to 25°C and stirred for 1 hour. After the reaction was completed, dichloromethane (20 mL) was added to the reaction solution at 0° C., water (10 mL) was added slowly to quench the reaction, and it was extracted with dichloromethane (30 mL*3). The organic phases were combined, dried over anhydrous sodium sulfate, and concentrated to obtain the target compound BB-1-8. LCMS, m/z=287.0 [M+1].
Step 7: Synthesis of compound BB-1
In a three-neck flask, at 0°C, to a tetrahydrofuran (50 mL) solution of BB-1-8 (1.9 g, 6.64 mmol, 1 eq) was added methyl magnesium bromide (3M, 5.53 mL, 2.5 eq). The reaction solution was stirred at 25°C for 5 hours under the protection of nitrogen. After the reaction was completed, at 0°C, water (10 mL) was slowly added to the reaction solution to quench the reaction, and then extracted with dichloromethane (10 mL*3). The organic phases were combined, dried with anhydrous sodium sulfate, concentrated, and subjected to preparative high performance liquid chromatography (column: Phenomenex Luna C18 200*40mm*10μm; mobile phase: [water (0.1%TFA)-acetonitrile]; B%: 15%- 35%, 10min) Purification (B is acetonitrile) to obtain BB-1.
Step 8: Synthesis of compound WX001-2
To the N,N-dimethylformamide (20mL) solution of WX001-1 (2g, 10.47mmol, 1eq) and potassium carbonate (4.34g, 31.41mmol, 3eq), add 2-iodopropane (3.56g, 20.94 mmol, 2.09mL, 2eq). The reaction solution was stirred at 90°C for 12 hours under the protection of nitrogen. After the reaction was completed, water (20 mL) was added to the reaction solution to quench the reaction, and then extracted with methyl tert-butyl ether (10 mL*3). The organic phases were combined, dried over anhydrous sodium sulfate, concentrated, and purified by column chromatography (petroleum ether: ethyl acetate = 5:1) to obtain the target compound WX001-2. 1 H NMR(400MHz, CDCl 3 )δ7.23(dd,J=2.4,10.6Hz,1H), 7.16(td,J=1.9,8.8Hz,1H), 6.85(t,J=8.7Hz,1H) , 4.49 (spt, J = 6.1 Hz, 1H), 1.35 (d, J = 6.1 Hz, 6H).
Step 9: Synthesis of compound WX001-3
To the dioxane (20mL) solution of WX001-2 (2g, 8.58mmol, 1eq), add double pinacol borate (2.40g, 9.44mmol, 1.1eq), potassium acetate (1.68g, 17.16 mmol, 2eq) and Pd(dppf)Cl 2 (627.87mg, 858.09μmol, 0.1eq). The reaction solution was stirred at 90°C for 3 hours under the protection of nitrogen. After the reaction was completed, water (20 mL) was added to the reaction solution to quench the reaction, and then extracted with dichloromethane (30 mL*3). The organic phases were combined, dried over anhydrous sodium sulfate, concentrated, and the crude product was purified by column chromatography (petroleum ether: ethyl acetate = 5:1) to obtain the target compound WX001-3. 1 H NMR (400MHz, CDCl 3 ) δ7.54-7.44 (m, 2H), 6.95 (t, J = 8.1 Hz, 1H), 4.60 (spt, J = 6.1 Hz, 1H), 1.37 (s, 3H) ,1.36(s,3H),1.33(s,12H).
Step 10: Synthesis of compound WX001-5
To WX001-3 (2.65g, 9.46mmol, 1eq) and WX001-4 (2.47g, 9.46mmol, 1eq) N,N-dimethylformamide/ethanol/water (265mL, volume ratio: 2/1/ 1) In the solution, add Pd(PPh 3 ) 4 (546.55 mg, 472.97 μmol, 0.05 eq) and sodium carbonate (3.01 g, 28.38 mmol, 3 eq). The reaction solution was stirred at 80°C for 12 hours under the protection of nitrogen. After the completion of the reaction, the reaction solution was filtered while hot (80℃) to obtain the mother liquor. After the mother liquor was spin-dried, dichloromethane (30mL) and water (30mL) were added. A large amount of insoluble matter was formed. After filtration, it was subjected to preparative high performance liquid chromatography. Purified to obtain the target compound WX001-5. LCMS, m/z=288.1 [M+1].
Step 11: Synthesis of compound WX001-6
To BB-1 (230mg, 760.90μmol, 1eq), WX001-5 (218.60mg, 760.90μmol, 1eq) and PPh 3 (299.36mg, 1.14mmol, 1.5eq) in tetrahydrofuran (50mL) solution at 25℃ , Add diisopropyl azodicarboxylate (230.79 mg, 1.14 mmol, 221.91 μL, 1.5 eq). The reaction solution was stirred at 45°C for 5 hours under the protection of nitrogen. After the completion of the reaction, the reaction solution was directly concentrated, and purified by a thin-layer chromatography plate (dichloromethane:methanol=15:1) to obtain an isomer mixture WX001-6.
Step 12: Synthesis of the compound of formula (I)
WX001-6 was purified by supercritical fluid chromatography (column: DAICEL CHIRALPAK AD-H (250mm*30mm, 5μm); mobile phase: [0.1% ammonia in ethanol]; B%: 22%-22%, 8min) (B It is 0.1% ammonia in ethanol) to obtain the compound of formula (I) (retention time is 2.29 min), and the structure of the compound of formula (I) is confirmed by a single crystal to be correct. 1 H NMR (400MHz, CD 3 OD) δ 8.95 (br s, 1H), 8.03 (s, 1H), 8.00-7.92 (m, 1H), 7.91-7.82 (m, 1H), 7.42-7.29 (m ,2H),7.27-7.09(m,1H),7.22(br t,J=8.6Hz,1H),6.96-6.71(m,2H),6.20(q,J=6.6Hz,1H),4.68(td , J=6.1, 11.9 Hz, 1H), 1.91 (d, J=7.0 Hz, 3H), 1.36 (d, J=5.7 Hz, 6H); LCMS, m/z=572.2 [M+1].
////////////TGR 1202
















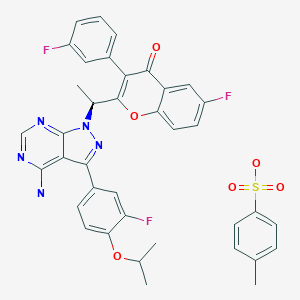
 Umbralisib tosylate
Umbralisib tosylate
 Structrual Formula Illustration")

 clip
First new chemical entity discovered by Indian scientists gets USFDA approval
clip
First new chemical entity discovered by Indian scientists gets USFDA approval
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
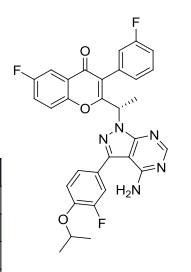



 TGR-1202.with Idelalisib and IPI-145 (left to right) for comparison.
TGR-1202.with Idelalisib and IPI-145 (left to right) for comparison.

 Example 1: Preparation of the PTSA Salt of TGR-1202 (Form A)
Example 1: Preparation of the PTSA Salt of TGR-1202 (Form A)
 7100 g of TGR-1202 was charged in a reactor containing 56.8 litres of acetone and stirred at ambient temperature. 4680 g of p-toluene sulphonic acid was added and the reaction mixture was heated at a temperature of 60-65° C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 142 litres of diethyl ether was then added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass. The solid mass was re-suspended in diethyl ether, stirred for 6 hours, and then filtered to yield a solid mass which was subsequently dissolved in 56.8 litres of acetone, filtered through a HiFlow bed, and concentrated under reduced pressure. The resulting residue mass was stirred with water overnight, then filtered and vacuum dried to yield 6600 g of the PTSA salt of TGR-1202. HPLC: 99.21% and chiral purity of 99.64:0.36 (S:R).
Example 2: Preparation of the PTSA Salt of TGR-1202 (Form B)
7100 g of TGR-1202 was charged in a reactor containing 56.8 litres of acetone and stirred at ambient temperature. 4680 g of p-toluene sulphonic acid was added and the reaction mixture was heated at a temperature of 60-65° C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 142 litres of diethyl ether was then added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass. The solid mass was re-suspended in diethyl ether, stirred for 6 hours, and then filtered to yield a solid mass which was subsequently dissolved in 56.8 litres of acetone, filtered through a HiFlow bed, and concentrated under reduced pressure. The resulting residue mass was stirred with water overnight, then filtered and vacuum dried to yield 6600 g of the PTSA salt of TGR-1202. HPLC: 99.21% and chiral purity of 99.64:0.36 (S:R).
Example 2: Preparation of the PTSA Salt of TGR-1202 (Form B)
 1000 g of TGR-1202 was charged in a reactor containing 8 litres of acetone and stirred at ambient temperature. 666 g of p-toluene sulphonic acid was then added and the reaction mixture was heated at a temperature of 60-65 °C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 20 litres of diethyl ether was added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass which was then vacuum dried to yield 1150 g of the PTSA salt of TGR-1202. HPLC: 99.33% and chiral purity: 99.61:0.39 (S:R).
Table 1 lists the XRPD pattern peaks and relative peak intensities for the products of Examples 1 and 2.
TABLE 1
1000 g of TGR-1202 was charged in a reactor containing 8 litres of acetone and stirred at ambient temperature. 666 g of p-toluene sulphonic acid was then added and the reaction mixture was heated at a temperature of 60-65 °C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 20 litres of diethyl ether was added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass which was then vacuum dried to yield 1150 g of the PTSA salt of TGR-1202. HPLC: 99.33% and chiral purity: 99.61:0.39 (S:R).
Table 1 lists the XRPD pattern peaks and relative peak intensities for the products of Examples 1 and 2.
TABLE 1
 The tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 2 exhibited a Cmax about 2.5 fold and an area under the curve (AUC) about 1.9 fold greater than that of the tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 1. The results are provided in Table 8 below.
TABLE 8
The tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 2 exhibited a Cmax about 2.5 fold and an area under the curve (AUC) about 1.9 fold greater than that of the tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 1. The results are provided in Table 8 below.
TABLE 8
 PATENT
WO 2014071125
http://www.google.com/patents/WO2014071125A1?cl=en
formula (A) that is a ΡΒΚδ selective inhibitor,
PATENT
WO 2014071125
http://www.google.com/patents/WO2014071125A1?cl=en
formula (A) that is a ΡΒΚδ selective inhibitor,









 B1 IS DESIRED
(S)-2- (l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-6- fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (compound-B l)
Intermediate 11
[119] Intermediate 11: 4-bromo-2-fluoro-l-isopropoxybenzene:To a solution of 4-bromo-2- fluorophenol (lOg, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8ml, 62.62 mmol) and triphenylphosphine (20.6g, 78.52 mmol) were added and heated to 45 C followed by diisopropylazodicarboxylate (15.4ml, 78 52 mmol). The mixture was refluxed for lh, concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a colourless liquid (13. lg, 99%) which was used without purification in the next step. Intermediate 12
[120] Intermediate 12: 2-(3-fluoro-4-isopropoxyphenyl)-4,4,5,5-tetramethyl- 1,3,2- dioxaborolane: Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15g, 58.96 mmol) were added to a solution of intermediate 11 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [1,1 ‘- Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH2Cl2 (4.4g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 13
[121] Intermediate 13: 3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- amine: To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF 110 ml), ethanol (55 ml) and water (55 ml), intermediate 12 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min. Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h, the reaction mixture was filtered though celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
Example Bl
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
[127] To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate ( 0.15 ml, 0.749 mmol) was added heated to 45°C. After 2h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 %). MP: 139- 142°C. Mass : 571.7 (M H-NMR (δ ppm, CDC13, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J = 8.2,3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J = 8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.11 (q, J = 7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J = 6.1 Hz, 1H), 2.00 (d, J = 7.1Hz, 3H), 1.42 (d, J = 6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64min.).
PATENT
US 2014/0011819 describe the synthesis of TGR-1202 (Example B l)
http://www.google.co.in/patents/US20140011819
Example B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
B1 IS DESIRED
(S)-2- (l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-6- fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (compound-B l)
Intermediate 11
[119] Intermediate 11: 4-bromo-2-fluoro-l-isopropoxybenzene:To a solution of 4-bromo-2- fluorophenol (lOg, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8ml, 62.62 mmol) and triphenylphosphine (20.6g, 78.52 mmol) were added and heated to 45 C followed by diisopropylazodicarboxylate (15.4ml, 78 52 mmol). The mixture was refluxed for lh, concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a colourless liquid (13. lg, 99%) which was used without purification in the next step. Intermediate 12
[120] Intermediate 12: 2-(3-fluoro-4-isopropoxyphenyl)-4,4,5,5-tetramethyl- 1,3,2- dioxaborolane: Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15g, 58.96 mmol) were added to a solution of intermediate 11 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [1,1 ‘- Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH2Cl2 (4.4g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 13
[121] Intermediate 13: 3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- amine: To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF 110 ml), ethanol (55 ml) and water (55 ml), intermediate 12 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min. Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h, the reaction mixture was filtered though celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
Example Bl
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
[127] To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate ( 0.15 ml, 0.749 mmol) was added heated to 45°C. After 2h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 %). MP: 139- 142°C. Mass : 571.7 (M H-NMR (δ ppm, CDC13, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J = 8.2,3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J = 8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.11 (q, J = 7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J = 6.1 Hz, 1H), 2.00 (d, J = 7.1Hz, 3H), 1.42 (d, J = 6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64min.).
PATENT
US 2014/0011819 describe the synthesis of TGR-1202 (Example B l)
http://www.google.co.in/patents/US20140011819
Example B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one

