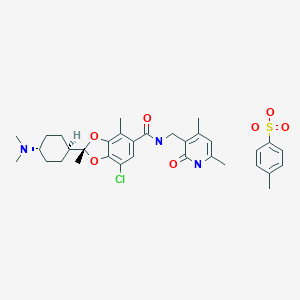
Valemetostat, also known as DS-3201 is a potent, selective and orally active EZH1/2 inhibitor. DS-3201 selectively inhibits the activity of both wild-type and mutated forms of EZH1 and EZH2. Inhibition of EZH1/2 specifically prevents the methylation of lysine 27 on histone H3 (H3K27). This decrease in histone methylation alters gene expression patterns associated with cancer pathways, enhances transcription of certain target genes, and results in decreased proliferation of EZH1/2-expressing cancer cells.
OriginatorDaiichi Sankyo Inc
DeveloperCALYM Carnot Institute; Daiichi Sankyo Inc; Lymphoma Academic Research Organisation; Lymphoma Study Association; University of Texas M. D. Anderson Cancer Center
ClassAmides; Amines; Antineoplastics; Benzodioxoles; Chlorinated hydrocarbons; Cyclohexanes; Pyridones; Small molecules
Mechanism of ActionEnhancer of zeste homolog 1 protein inhibitors; Enhancer of zeste homolog 2 protein inhibitors
Orphan Drug StatusYes – Adult T-cell leukaemia-lymphoma; Peripheral T-cell lymphoma
The equipment and measurement conditions for the powder X-ray diffraction measurement in the examples are as follows.
Model: Rigaku Rint TTR-III
Specimen: Appropriate
X-ray generation conditions: 50 kV, 300 mA
Wavelength: 1.54 Å (Copper Kα ray)
Measurement temperature: Room temperature
Scanning speed: 20°/min
Scanning range: 2 to 40°
Sampling width: 0.02°
[0043]
(Reference Example 1) Production of ethyl trans-4-[(tert-butoxycarbonyl)amino]cyclohexanecarboxylate
[0044]
[hua 6]
[0045]
Under a nitrogen atmosphere, ethanol (624 L) and ethyl trans-4-aminocyclohexanecarboxylate monohydrochloride (138.7 kg, 667.8 mol) were added to a reaction vessel and cooled. Triethylamine (151.2 kg, 1495 .5 mol) and di-tert-butyl dicarbonate (160.9 kg, 737.2 mol) were added dropwise while maintaining the temperature below 20°C. After stirring at 20-25°C for 4 hours, water (1526 kg) was added dropwise at 25°C or lower, and the mixture was further stirred for 2 hours. The precipitated solid was collected by filtration, washed with a mixture of ethanol:water 1:4 (500 L), and dried under reduced pressure at 40°C to obtain 169.2 kg of the title compound (yield 93.4%). . 1 H NMR (500 MHz, CDCl 3 ): δ 4.37 (br, 1H), 4.11 (q, J = 2.8 Hz, 2H), 3.41 (br, 1H), 2.20 (tt, J = 4.8, 1.4 Hz, 1H),2.07(m,2H),2.00(m,2H),1.52(dq,J=4.6,1.4Hz,2H),1.44(s,9H),1.24(t,J=2.8Hz,3H), 1.11(dq,J=4.6,1.4Hz,2H)
[0046]
(Reference Example 2) Production of tert-butyl = [trans-4-(hydroxymethyl)cyclohexyl]carbamate
[0047]
[hua 7]
[0048]
Under a nitrogen atmosphere, tetrahydrofuran (968 kg), ethyl = trans-4-[(tert-butoxycarbonyl)amino]cyclohexanecarboxylate (110 kg, 405.4 mol), lithium chloride (27.5 kg, 648 kg) were placed in a reaction vessel. .6 mol), potassium borohydride (32.8 kg, 608.1 mol), and water (2.9 L, 162.2 mol) were added, the temperature was slowly raised to 50°C, and the mixture was further stirred for 6 hours. Cooled to 0-5°C. Acetone (66 L) and 9 wt % ammonium chloride aqueous solution (1210 kg) were added dropwise while maintaining the temperature at 20° C. or lower, and the mixture was stirred at 20-25° C. for 1 hour. Additional ethyl acetate (550 L) was added, the aqueous layer was discarded and the organic layer was concentrated to 550 L. Ethyl acetate (1650 L) and 9 wt% aqueous ammonium chloride solution (605 kg) were added to the residue, and the aqueous layer was discarded after stirring. Washed sequentially with water (550 L). The organic layer was concentrated to 880 L, ethyl acetate (660 L) was added to the residue, and the mixture was concentrated to 880 L while maintaining the internal temperature at 40-50°C. The residue was cooled to 0-5° C. and stirred for 1 hour, petroleum ether (1760 L) was added dropwise over 30 minutes, and the mixture was stirred at the same temperature for 2 hours. The precipitated solid was collected by filtration, washed with a petroleum ether:ethyl acetate 3:1 mixture (220 L) cooled to 0-5°C, and dried at 40°C under reduced pressure to give 86.0 kg of the title compound (yield: obtained at a rate of 92.3%). 1 H NMR (500 MHz, CDCl 3 ): δ 4.37 (br, 1H), 3.45 (d, J = 2.2 Hz, 2H), 3.38 (br, 1H), 2.04 (m, 2H),
1.84(m,2H),1.44(m,10H),1.28-1.31(m,1H),1.00-1.13(m,4H)
[0049]
(Reference Example 3) Production of tert-butyl = [trans-4-(2,2-dibromoethenyl)cyclohexyl]carbamate
[0050]
[hua 8]
[0051]
(Step 1)
Under a nitrogen atmosphere, ethyl acetate (50 L), tert-butyl = [trans-4-(hydroxymethyl)cyclohexyl]carbamate (2.5 kg, 10.90 mol), potassium bromide ( 39.3 g, 0.33 mol), 2,2,6,6-tetramethylpiperidine 1-oxyl (51.1 g, 0.33 mol), 4.8% aqueous sodium hydrogen carbonate solution (26.25 kg ) was added and cooled to 0-5°C, 9.9% sodium hypochlorite (8.62 kg, 11.45 mol) was added at 5°C or lower, and the mixture was further stirred at 0°C for 4 hours. Sodium sulfite (250 g) was added to the mixture and stirred at 0-5°C for 30 minutes before warming to 20-25°C. Thereafter, the aqueous layer was discarded and washed with a 20% aqueous sodium chloride solution (12.5 kg), then the organic layer was dried over sodium sulfate and concentrated to 7.5 L. Ethyl acetate (12.5 L) was added to the residue, the mixture was concentrated again to 7.5 L, and used in the next reaction as a tert-butyl=(trans-4-formylcyclohexyl)carbamate solution.
[0052]
(Step 2)
Under a nitrogen atmosphere, tetrahydrofuran (30 L) and triphenylphosphine (5.72 kg, 21.8 mol) were added to a reaction vessel, heated to 40°C, and stirred for 5 minutes. Carbon tetrabromide (3.61 kg, 10.9 mol) was added over 30 minutes and stirred at 40-45° C. for another 30 minutes. A mixture of tert-butyl (trans-4-formylcyclohexyl)carbamate solution and triethylamine (2.54 kg, 25.1 mol) was added below 45°C over 20 minutes and stirred at 40°C for an additional 15 hours. After cooling the reaction solution to 0° C., water (0.2 L) was added at 10° C. or lower, and water (25 L) was added. After heating to 20-25° C., the aqueous layer was discarded, ethyl acetate (4.5 kg) and 10% aqueous sodium chloride solution (25 kg) were added, and after stirring, the aqueous layer was discarded again. After the obtained organic layer was concentrated to 15 L, 2-propanol (19.65 kg) was added and concentrated to 17.5 L. 2-Propanol (11.78 kg) and 5 mol/L hydrochloric acid (151.6 g) were added to the residue, and the mixture was stirred at 25-35°C for 2.5 hours. Water (16.8 L) was added dropwise to the resulting solution, and the mixture was stirred at 20-25°C for 30 minutes and then stirred at 0°C for 2 hours. The precipitated solid was collected by filtration, washed with a mixture (11 kg) of acetonitrile:water 60:40 cooled to 0-5°C, and dried at 40°C under reduced pressure to give 3.05 kg of the title compound (yield 73%). .0%). 1 H NMR (500 MHz, CDCl3):δ6.20(d,J=3.6Hz,1H),4.37(br,1H),3.38(br,1H),2.21(dtt,J=3.6,4.6,1.4Hz,1H),2.05-2.00(m,2H),1.80-1.83(m,2H),1.44(s,9H),1.23(ddd,J=9.9,5.3,1.2 Hz,2H), 1.13(ddt,J=4.6,1.4,5.2 Hz,2H)
[0053]
(Reference Example 4) Production of tert-butyl = (trans-4-ethynylcyclohexyl) carbamate
[0054]
[Chemical 9]
[0055]
Under a nitrogen atmosphere, toluene (1436 kg), tert-butyl = [trans-4-(2,2-dibromoethenyl)cyclohexyl]carbamate (110 kg, 287.1 mol), and N,N,N ‘,N’-Tetramethylethane-1,2-diamine (106.7 kg, 918.8 mol) was added and cooled to -10°C. An isopropylmagnesium chloride-tetrahydrofuran solution (2.0 mol/L, 418 kg, 863 mol) was added dropwise at -5°C or lower, and stirred at -10°C for 30 minutes. After the reaction, 5 mol/L hydrochloric acid (465 kg) was added at 5°C or lower, heated to 20-25°C, and further 5 mol/L hydrochloric acid (41.8 kg) was used to adjust the pH to 5.0-. adjusted to 6.0. After discarding the aqueous layer, the organic layer was washed twice with water (550 L) and concentrated to 550 L. 2-Propanol (1296 kg) was added to the concentrate and concentrated to 550 L again. Further, 2-propanol (1296 kg) was added to the residue, and after concentrating to 550 L, water (770 L) was added dropwise in 4 portions. At that time, it was stirred for 30 minutes after each addition. After the addition, the mixture was stirred for 1 hour and further stirred at 0° C. for 1 hour. The precipitated solid was collected by filtration, washed with a 5:7 mixture of 2-propanol:water (550 L) cooled to 0-5°C, and dried at 40°C under reduced pressure to yield 57.8 kg of the title compound. obtained at a rate of 90.2%). 1 H NMR (500 MHz, CDCl 3 ): δ 4.36 (br, 1H), 3.43 (br, 1H), 2.18-2.23 (m, 1H), 1.97-2.04 (m, 5H), 1.44-1.56 (m, 11H ),1.06-1.14(m,2H)
[0056]
(Reference Example 5) Production of 4,6-dimethyl-2-oxo-1,2-dihydropyridine-3-carbonitrile
[0057]
[Chemical 10]
[0058]
Under a nitrogen atmosphere, water (300 L), 2-cyanoacetamide (20 kg, 238 mol), 1-pentane-2-4-dione (26.2 kg, 262 mol), potassium carbonate (3.29 mol) were added to a reaction vessel. kg, 23.8 mol) was added and stirred at room temperature for 6 hours or longer. After the reaction, the precipitated solid was collected by filtration, washed with water (60 L), further washed with a mixture of methanol (40 L) and water (40 L), and dried under reduced pressure at 40°C to give the title compound as 34 Obtained in .3 kg (97.3% yield). 1 H NMR (500 MHz, DMSO-d 6 ): δ 2.22 (s, 3H), 2.30 (s, 3H), 6.16 (s, 1H), 12.3 (brs, 1H)
[0059]
(Reference Example 6) Production of 3-(aminomethyl)-4,6-dimethylpyridin-2(1H)-one monohydrochloride
[0060]
[Chemical 11]
[0061]
Under a nitrogen atmosphere, water (171 L), methanol (171 L), 4,6-dimethyl-2-oxo-1,2-dihydropyridine-3-carbonitrile (17.1 kg, 116 mol), concentrated After adding hydrochloric acid (15.8 kg, 152 mol) and 5% palladium carbon (55% wet) (3.82 kg), the inside of the reaction vessel was replaced with hydrogen. Then, the mixture was pressurized with hydrogen and stirred overnight at 30°C. After the reaction, the reaction vessel was purged with nitrogen, the palladium on carbon was removed by filtration, and the palladium on carbon was washed with a 70% aqueous solution of 2-propanol (51 L). Activated carbon (0.86 kg) was added to the filtrate and stirred for 30 minutes. Activated carbon was removed by filtration and washed with 70% aqueous 2-propanol solution (51 L). The filtrate was concentrated under reduced pressure until the liquid volume became 103 L, and 2-propanol (171 L) was added. The mixture was again concentrated under reduced pressure until the liquid volume reached 103 L, then 2-propanol (171 L) was added, and the mixture was stirred for 1 hour or longer. Precipitation of a solid was confirmed, and the solution was concentrated to a volume of 103 L. Further, 2-propanol (51 L) was added, and after concentration under reduced pressure until the liquid volume reached 103 L, the mixture was stirred at 50° C. for 30 minutes. After adding acetone (171 L) over 1 hour while keeping the internal temperature at 40° C. or higher, the mixture was stirred at 40 to 45° C. for 30 minutes. The solution was cooled to 25°C and stirred for 2 hours or longer, and the precipitated solid was collected by filtration, washed with acetone (86 L) and dried under reduced pressure at 40°C to give 19.7 kg of the title compound (yield 90.4%). ). 1 H NMR (500 MHz, methanol-d 4 ): δ 2.27 (s, 3H), 2.30 (s, 3H), 4.02 (s, 2H), 6.16 (s, 1H)
[0062]
(Example 1-1) Production of methyl 5-chloro-3,4-dihydroxy-2-methylbenzoate
[0063]
[Chemical 12]
[0064]
Under a nitrogen atmosphere, water (420 L), toluene (420 L), acetonitrile (420 L), and methyl 3,4-dihydroxy-2-methylbenzoate (1) (60 kg, 329 mol) were added to the reactor and cooled. After that, sulfuryl chloride (133.4 kg, 988 mol) was added dropwise while maintaining the temperature at 20°C or lower. After the reaction, the mixture was separated into an organic layer 1 and an aqueous layer, acetonitrile (60 L) and toluene (120 L) were added to the aqueous layer, and the mixture was stirred. Water (420 L) and acetonitrile (210 L) were added to the organic layer 1, and after cooling, sulfuryl chloride (88.9 kg, 659 mol) was added dropwise at 20°C or lower, and sulfuryl chloride (53.2 kg, 394 mol) was added. ) was added in portions. After the reaction, the mixture was separated into an organic layer 3 and an aqueous layer, and the organic layer 2 was added to the aqueous layer and stirred. Water (420 L), acetonitrile (210 L) were added to the combined organic layer, sulfuryl chloride (44.5 kg, 329 mol) was added dropwise below 20°C, and sulfuryl chloride (106.4 kg, 788 mol) was added. ) was added in portions. After the reaction, the organic layer 4 and the aqueous layer were separated, acetonitrile (60 L) and toluene (120 L) were added to the aqueous layer, and the mixture was stirred. The combined organic layers were washed three times with 20 wt % aqueous sodium chloride solution (300 L) and then concentrated under reduced pressure to 600 L. After repeating the operation of adding toluene (300 L) and concentrating under reduced pressure to 600 L again twice, the mixture was heated and stirred at 60° C. for 1 hour. After cooling to room temperature, the precipitated solid was collected by filtration, washed with toluene (120 L), and dried under reduced pressure at 40°C to give 52.1 kg of the crude title compound (2) (yield: 73.0%). ).
[0065]
Under a nitrogen atmosphere, toluene (782 L) and crude title compound (52.1 kg, 241 mol) were added to a reactor and heated to 80°C. After confirming that the crystals were completely dissolved, they were filtered and washed with heated toluene (261 L). The mixture was cooled to 60° C. and stirred for 0.5 hours after crystallization. After cooling to 10°C, the precipitated solid was collected by filtration, washed with toluene (156 L), and dried under reduced pressure at 40°C to give 47.9 kg of the title compound (2) (yield 91.9%). Acquired. 1 H NMR (500 MHz, methanol-d 4 ): δ 2.41 (s, 3H), 3.82 (s, 3H), 7.41 (s, 1H)
[0066]
(Example 1-2) Examination of chlorination conditions 1 Since
it is difficult to remove compound (1), which is the starting material, and compound (4), which is a by-product of the reaction, even in subsequent steps, need to control. Therefore, chlorination was investigated in the same manner as in Example 1-1 using compound (1) as a starting material. Table 1 shows the results.
[0067]
[Chemical 13]
[0068]
[Table 1]
[0069]
HPLC condition
detection: 220 nm
column: ACQUITY UPLC BEH C18 (2.1 mm ID x 50 mm, 1.7 μm, Waters)
column temperature: 40 ° C
mobile phase: A: 0.1 vol% trifluoroacetic acid aqueous solution, B: acetonitrile
Gradient conditions:
(Example 1-3) Examination of chlorination conditions 2
Compound (1) was used as a starting material, sulfuryl chloride was used as a chlorination reagent, and chlorination in various solvents was examined. Table 3 shows the results.
[0073]
[table 3]
[0074]
(Example 2) Methyl (2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5- Manufacture of carboxylates
[0075]
[Chemical 14]
[0076]
Toluene (9.0 L), tert-butyl = (trans-4-ethynylcyclohexyl) carbamate (2.23 kg, 9.99 mol), methyl = 5-chloro-3,4- were added to a reaction vessel under a nitrogen atmosphere. Dihydroxy-2-methylbenzoate (1.80 kg, 8.31 mol), tri(o-tolyl)phosphine (76.0 g, 250 mmol), triruthenium dodecacarbonyl (53.0 g, 82.9 mmol) ) was added, and the mixture was heated and stirred at 80 to 90° C. for 7 hours under an oxygen-containing nitrogen stream. The reaction solution was cooled to room temperature to obtain a toluene solution of the title compound.
[0077]
(Example 3) (2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carvone acid production
[0078]
[Chemical 15]
[0079]
Methyl = (2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole obtained in Example 2 -5-carboxylate toluene solution (13 L, equivalent to 7.83 mol), methanol (9.0 L), 1,2-dimethoxyethane (3.6 L), 5 mol / L sodium hydroxide aqueous solution ( 2.50 L, 12.5 mol) was added and stirred at 55-65° C. for 3 hours. After adding water (5.4 L), the mixture was allowed to stand and separated into an organic layer and an aqueous layer. After cooling to room temperature, 1,2-dimethoxyethane (16.2 L) was added to the aqueous layer, and after adjusting the pH to 4.0 to 4.5 with 3 mol/L hydrochloric acid, toluene (5.4 L) was added. added. After heating to 50-60° C., the organic layer and aqueous layer were separated, and the organic layer was washed with a 20 wt % sodium chloride aqueous solution (7.2 L). Then, 1,2-dimethoxyethane (21.6 L) was added to the organic layer, and after concentration under reduced pressure to 9 L, 1,2-dimethoxyethane (21.6 L) was added and heated to 50-60°C. After that, filtration was performed to remove inorganic substances. Then, after washing with 1,2-dimethoxyethane (1.8 L), the 1,2-dimethoxyethane solution of the title compound (quantitative value 89.6% (Example 2 total yield from ), corresponding to 7.45 mol).
[0080]
(Example 4) (1S)-1-phenylethanaminium (2R)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1, Preparation of 3-benzodioxole-5-carboxylate
[0081]
[Chemical 16]
[0082]
(2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5 obtained in Example 3 – A solution of carboxylic acid in dimethoxyethane (21.6 L, corresponding to 7.45 mol) was heated to 75-80°C, and then (1S)-1-phenylethanamine (1.02 kg, 8.42 mmol). was added and stirred for 4 hours. A mixture of 1,2-dimethoxyethane (9.2 L) and water (3.4 L) heated to 50-60° C. was added, stirred, and then cooled to room temperature. The precipitated solid was collected by filtration and washed with 1,2-dimethoxyethane (9 L) to give a crude title compound (1.75 kg (converted to dry matter), yield 38.5% (Example 2 total yield from ) and an optical purity of 93.8% ee).
[0083]
Under a nitrogen atmosphere, a 1,2-dimethoxyethane aqueous solution (13.6 L) was placed in a reaction vessel, and (1S)-1-phenylethanaminium obtained in step 1 (2R)-2-{trans-4-[(tert -Butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carboxylate crude (1.70 kg equivalent, 3.11 mol) was added. After that, 5 mol/L hydrochloric acid (0.56 L, 2.8 mol) was added dropwise. After stirring at room temperature for 10 minutes or longer, the mixture was heated to 75° C. or higher, and (1S)-1-phenylethanamine (360 g, 2.97 mmol) was dissolved in 1,2-dimethoxyethane (2.6 L). The solution was added dropwise over 1 hour. It was then washed with 1,2-dimethoxyethane (0.9 L), stirred for 2 hours and cooled to 0-5°C. The slurry was collected by filtration and washed with 1,2-dimethoxyethane (5.1 L) cooled to 0-5° C. to give the title compound (1.56 kg, yield 91.9%, obtained with an optical purity of 99.5% ee). 1 H NMR (500 MHz, methanol-d 4 ): δ 1.15-1.23(m,2H), 1.28-1.35(m,2H), 1.42(s,9H),
1.59(s,3H), 1.60-1.61(d ,3H,J=7.0Hz,3H),1.80-1.86(dt,J=12.0,3.0Hz,1H),1.95-1.96(m,4H),2.27(s,3H),3.24-3.28(m,1H ),4.39-4.43(q,J=7.0Hz,1H),7.07(s,1H),7.37-7.45(m,5H)
[0084]
(Example 5) (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxylic acid monohydrochloride Manufacturing A
[0085]
[Chemical 17]
[0086]
(Step 1)
Under a nitrogen atmosphere, 1,2-dimethoxyethane (200 L) and (1S)-1-phenylethanaminium (2R)-2-{trans-4-[(tert-butoxycarbonyl) were placed in a reaction vessel. Amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carboxylate (equivalent to 87.64 kg, 160 mol), 35% hydrochloric acid (16.7 kg, 160 mol) was added and heated to 45-55° C., 35% hydrochloric acid (36.7 kg, 352 mol) was added dropwise in 7 portions and stirred for 3 hours after dropping. After cooling to room temperature, the reaction solution was added to a mixture of water (982 L) and 5 mol/L sodium hydroxide (166.34 kg, 702 mol). 3 mol/L hydrochloric acid (22.4 kg) was added dropwise to the resulting solution at 30°C, crystal precipitation was confirmed, and the mixture was stirred for 30 minutes or more, cooled to 10°C, and further stirred for 2 hours. After stirring, 3 mol/L hydrochloric acid (95.1 kg) was added dropwise at 10°C to adjust the pH to 7.0. The slurry liquid was collected by filtration, washed with water (293 L) cooled to 10° C., and (2R)-2-(trans-4-aminocyclohexyl)-7-chloro-2,4-dimethyl-1,3- Benzodioxol-5-carboxylic acid trihydrate was obtained (57.63 kg (converted to dry matter), yield 94.7%). 1 H NMR (500 MHz, methanol- d4 + D2O): 1.32-1.44 ( m, 4H), 1.61 (s, 3H), 1.89-1.94 (m, 1H), 2.01-2.13 (m, 4H) ,2.27(s,3H),2.99-3.07(m,1H),7.06(s,3H)
[0087]
(Step 2)
Under nitrogen atmosphere, 1,2-dimethoxyethane (115 L), (2R)-2-(trans-4-aminocyclohexyl)-7-chloro-2,4-dimethyl-1,3 -benzodioxole-5-carboxylic acid trihydrate (57.63 kg equivalent, 152 mmol), formic acid (34.92 kg, 759 mol), 37% formaldehyde aqueous solution (93.59 kg, 1153 mol) was added and stirred at 55-65°C for 2 hours. Cool to room temperature, add 2-propanol (864 L) and concentrate to 576 L under reduced pressure. 2-Propanol (231 L) was added thereto and concentrated under reduced pressure to 576 L. Further, 2-propanol (231 L) was added and concentrated under reduced pressure to 576 L. After concentration, 35% hydrochloric acid (20.40 kg, 196 mol) was added dropwise over 2 hours and stirred at room temperature for 30 minutes. Ethyl acetate (576 L) was added to the resulting slurry over 30 minutes and concentrated to 692 L. Ethyl acetate (461 L) was added followed by further concentration to 519 L. Ethyl acetate (634 L) was added to the residue and the mixture was stirred at room temperature for 2 hours. The precipitated solid was collected by filtration, washed with ethyl acetate (491 L) and dried under reduced pressure at 40°C to give the title compound (51. 56 kg, 87.1% yield). 1 H NMR (500 MHz, methanol-d 4 ): δ 1.38-1.47 (m, 2H), 1.53-1.61 (m, 2H), 1.67 (s, 3H), 1.99-2.05 (m, 1H), 2.13 -2.18(m,4H),2.38(s,3H),2.84(s,6H),3.19-3.25(dt,J=12.5,3.5Hz,1H),
7.53(s,1H)
[0088]
(Example 6) (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxylic acid monohydrochloride Manufacturing B
[0089]
[Chemical 18]
[0090]
Under a nitrogen atmosphere, formic acid (20 mL), 37% formaldehyde aqueous solution (15 mL), dimethoxyethane (10 mL), (1S)-1-phenylethanaminium (2R)-2-{trans-4- [(tert-Butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carboxylate (10 g, 18.3 mmol) was added and Stirred for 10 hours. After cooling to room temperature and filtering the insolubles, 2-propanol (100 mL) was added and the mixture was concentrated under reduced pressure until the liquid volume became 30 mL. While stirring at room temperature, ethyl acetate (120 mL) and concentrated hydrochloric acid (6.1 mL) were added to form a slurry. This was concentrated under reduced pressure to 30 mL, ethyl acetate (120 mL) was added, and then concentrated under reduced pressure to 30 mL again. After adding ethyl acetate (120 mL), the precipitated solid was collected by filtration, washed with ethyl acetate (50 mL) and dried under reduced pressure at 40°C to give 6.56 g of the title compound (yield 92.0%). Acquired.
[0091]
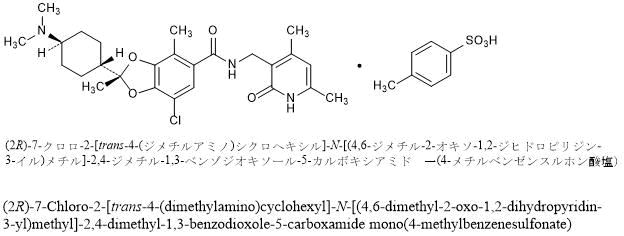
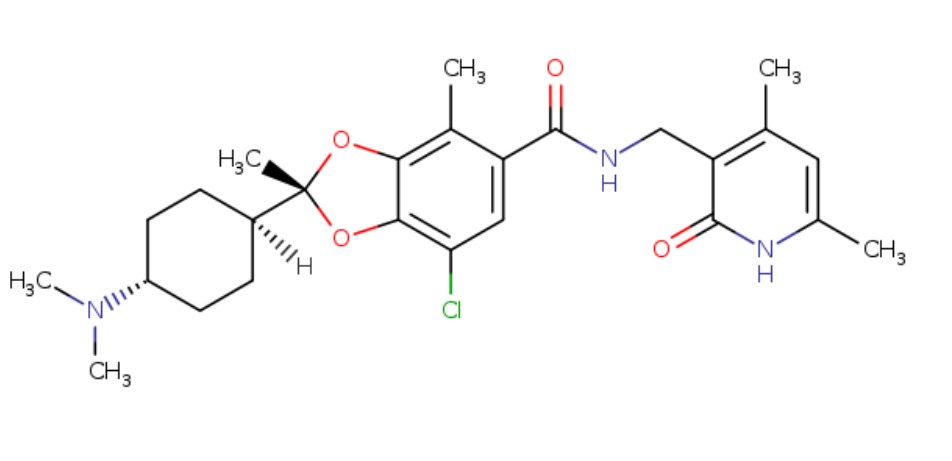
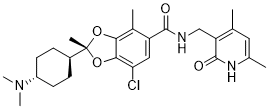
(Example 7) (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-N-[(4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl ) Preparation of methyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxamide p-toluenesulfonate
[0092]
[Chemical 19]
[0093]
Under nitrogen atmosphere, acetone (6.5 L), purified water (1.3 L), (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-2,4- Dimethyl-1,3-benzodioxole-5-carboxylic acid monohydrochloride (650.4 g, 1.67 mol), 3-(aminomethyl)-4,6-dimethylpyridin-2(1H)-one Monohydrochloride (330.1 g, 1.75 mol) and triethylamine (337 g, 3.33 mol) were added and stirred at room temperature for 30 minutes. After that, 1-hydroxybenzotriazole monohydrate (255 g, 1.67 mol), 1-ethyl-3-(dimethylaminopropyl)carbodiimide hydrochloride (383 g, 2.00 mmol) were added, and the mixture was stirred overnight at room temperature. Stirred. After adjusting the pH to 11 with 5 mol/L sodium hydroxide, toluene (9.8 L) was added, and after stirring, the mixture was separated into an organic layer 1 and an aqueous layer. Toluene (3.3 L) was added to the aqueous layer, and after stirring, the aqueous layer was discarded, and the obtained organic layer was combined with the previous organic layer 1. The combined organic layers were concentrated under reduced pressure to 9.75 L, toluene (6.5 L) was added and washed twice with purified water (3.25 L). The resulting organic layer was concentrated under reduced pressure to 4.875 L and 2-propanol (1.625 L) was added. A solution of p-toluenesulfonic acid monohydrate (0.12 kg, 0.631 mol) dissolved in 4-methyl-2-pentanone (1.14 L) was added to the organic layer heated to 68°C. The mixture was added dropwise over 5 hours and stirred at 68°C for 30 minutes. Furthermore, a solution of p-toluenesulfonic acid monohydrate (0.215 kg, 1.13 mol) dissolved in 4-methyl-2-pentanone (2.11 L) was added dropwise over 3.5 hours, Stirred at 68° C. for 30 minutes. After that, 4-methyl-2-pentanone (6.5 L) was added dropwise over 1 hour. After cooling to room temperature, the precipitated solid was collected by filtration, washed with 4-methyl-2-pentanone (3.25 L) and dried under reduced pressure at 40°C to give 1.035 kg of the crude title compound (yield 94%). .2%).
[0094]
Under a nitrogen atmosphere, 2-propanol (6.65 L) and the obtained crude title compound (950 g) were added to the reactor and stirred. Purified water (0.23 L) was added to completely dissolve the solid at 68° C., filtered, and washed with warm 2-propanol (0.95 L). After confirming that the solid was completely dissolved at an internal temperature of 68°C, the solution was cooled to 50°C. After cooling, seed crystals* (9.5 g, 0.01 wt) were added and stirred at 50° C. overnight. tert-Butyl methyl ether (11.4 L) was added dropwise thereto in 4 portions over 30 minutes each. At that time, it was stirred for 30 minutes after each addition. After cooling to room temperature, the precipitated solid was collected by filtration, washed with a mixture of 2-propanol (0.38 L) and tert-butyl methyl ether (3.42 L), and further treated with tert-butyl methyl ether (4.75 L). ) and dried under reduced pressure at 40° C. to obtain the title compound (915.6 g, yield 96.4%). 1 H NMR (500 MHz, methanol-d 4 ): δ 1.35-1.43 (m, 2H), 1.49-1.57 (m, 2H), 1.62 (s, 3H),
1.94-2.00 (dt, J = 12.5, 3.0Hz ,1H),2.09-2.13(m,4H),2.17(s,3H),2.24(s,3H),2.35(s,3H),2.36(s,3H),2.82(s,6H),3.16- 3.22(dt,J=12.0,3.5Hz,1H),4.42(s,2H),
6.10(s,1H),6.89(s,1H),7.22-7.24(d,J=8.0Hz,2H),7.69 -7.71(dt,J=8.0,1.5 Hz,2H)
*Seed crystal preparation method
Under a nitrogen atmosphere, 2-propanol (79.0 L) and the obtained crude title compound (7.90 kg) were added to a reactor and stirred. Purified water (7.9 L) was added to completely dissolve the solid, and activated carbon (0.40 kg) was added and stirred. After filtering the activated carbon, it was washed with 2-propanol (79.0 L) and concentrated to 58 L. 2-Propanol (5 L) was added to the residue, and after heating to 64° C., tert-butyl methyl ether (19.8 L) was added, and after crystal precipitation was confirmed, tert-butyl methyl ether (75. 1 L) was added in three portions. At that time, it was stirred for 30 minutes after each addition. After cooling to room temperature, the precipitated solid was collected by filtration, washed with a mixture of 2-propanol (7.9 L) and tert-butyl methyl ether (15.8 L), and dried under reduced pressure at 40°C to obtain seed crystals. The title compound was obtained (7.08 kg, 89.6% yield).
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT