














RNA, (Um-sp-(2′-deoxy-2′-fluoro)C-sp-Um-Um-Gm-(2′-deoxy-2′-fluoro)G-Um-Um-(2′-deoxy-2′-fluoro)A-Cm-Am-Um-Gm-(2′-deoxy-2′-fluoro)A-Am-(2′-deoxy-2′-fluoro)A-Um-Cm-Cm-Cm-Am-sp-Um-sp-Cm), complex with RNA (Um-sp-Gm-sp-Gm-Gm-Am-Um-(2′-deoxy-2′-fluoro)U-Um-(2′-deoxy-2′-fluoro)C-(2′-deoxy-2′-fluoro)A-(2′-deoxy-2′-fluoro)U-Gm-Um-Am-Am-Cm-Cm-Am-Am-Gm-Am) 3′-[[(2S,4R)-1-[29-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-14,14-bis[[3-[[3-[[5-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-1-oxopentyl]amino]propyl]amino]-3-oxopropoxy]methyl]-1,12,19,25-tetraoxo-16-oxa-13,20,24-triazanonacos-1-yl]-4-hydroxy-2-pyrrolidinyl]methyl hydrogen phosphate] (1:1)
Vutrisiran Sodium
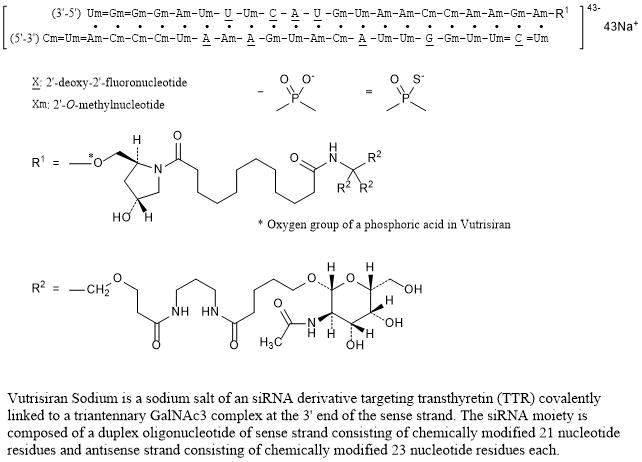
Nucleic Acid Sequence
Sequence Length: 44, 23, 2113 a 9 c 8 g 14 umultistranded (2); modified
Vutrisiran sodium
- ALN 65492
- Votrisiran
C530H672F9N171Na43O323P43S6 : 17289.77
[1867157-35-4 , Vutrisiran]
| Formula |
C530H672F9N171O323P43S6.43Na OR
C530H672F9N171Na43O323P43S6
|
|---|---|
| CAS |
1867157-35-4 , VURISIRAN
|
| Mol weight |
17289.7661
|
FDA APPROVED, AMVUTTRA, 2022/6/13
|
ブトリシランナトリウム
|
| Efficacy |
Gene expression regulator
|
|---|---|
| Disease |
Polyneuropathy of hereditary transthyretin-mediated amyloidosis [D
|
| Comment |
RNA interference (RNAi) drug
Treatment of transthyretin (TTR)-mediated amyloidosis (ATTR amyloidosis) |
UNII28O0WP6Z1P UNII
Vutrisiran
Vutrisiran Sodium is a sodium salt of an siRNA derivative targeting transthyretin (TTR) covalently linked to a triantennary GalNAc3 complex at the 3’ end of the sense strand. The siRNA moiety is composed of a duplex oligonucleotide of sense strand consisting of chemically modified 21 nucleotide residues and antisense strand consisting of chemically modified 23 nucleotide residues each.
Vutrisiran is a double-stranded small interfering ribonucleic acid (siRNA) that targets wild-type and mutant transthyretin (TTR) messenger RNA (mRNA).7 This siRNA therapeutic is indicated for the treatment of neuropathies associated with hereditary transthyretin-mediated amyloidosis (ATTR), a condition caused by mutations in the TTR gene.2 More than 130 TTR mutations have been identified so far,3 but the most common one is the replacement of valine with methionine at position 30 (Val30Met).2 The Val30Met variant is the most prevalent among hereditary ATTR patients with polyneuropathy, especially in Portugal, France, Sweden, and Japan.2
TTR mutations lead to the formation of misfolded TTR proteins, which form amyloid fibrils that deposit in different types of tissues. By targeting TTR mRNA, vutrisiran reduces the serum levels of TTR.6,7 Vutrisiran is commercially available as a conjugate of N-acetylgalactosamine (GalNAc), a residue that enables the delivery of siRNA to hepatocytes.5,7 This delivery platform gives vutrisiran high potency and metabolic stability, and allows for subcutaneous injections to take place once every three months.8 Another siRNA indicated for the treatment of polyneuropathy associated with hereditary ATTR is patisiran.2 Vutrisiran was approved by the FDA in June 2022.
CLIP
https://www.nature.com/articles/s41392-020-0207-x
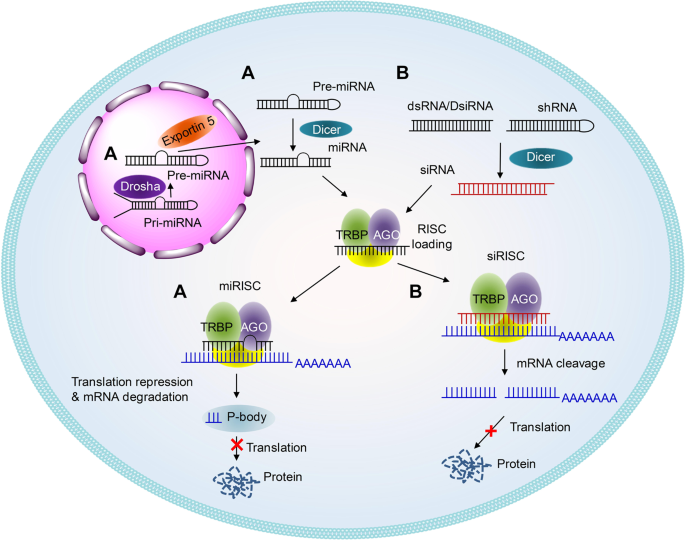
Schematic illustrations of the working mechanisms of miRNA (a) and siRNA (b)
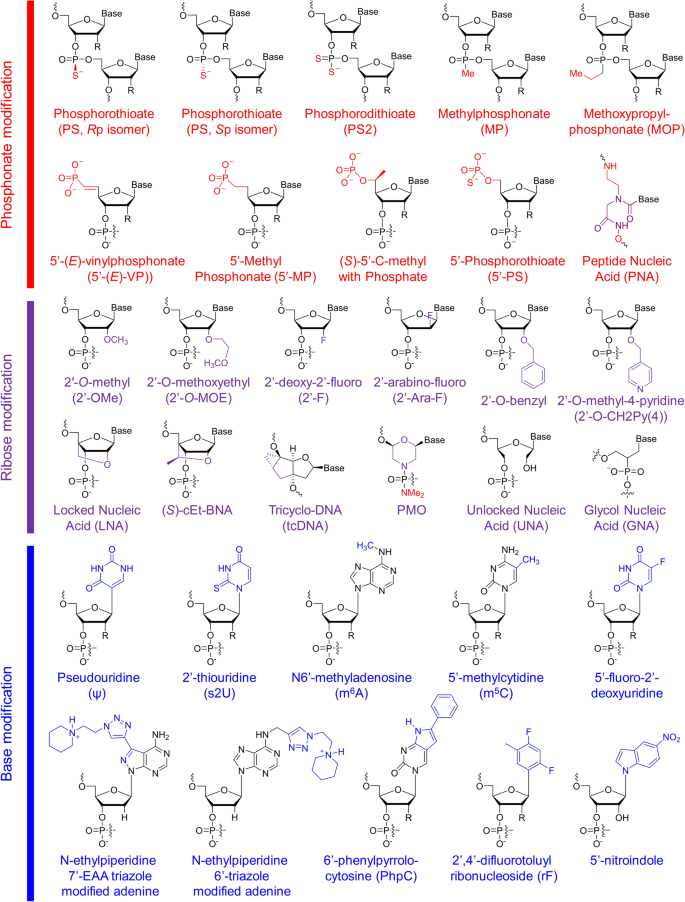
Structures of chemical modifications and analogs used for siRNA and ASO decoration. According to the modification site in the nucleotide acid, these structures can be divided into three classes: phosphonate modification, ribose modification and base modification, which are marked in red, purple and blue, respectively. R = H or OH, for RNA or DNA, respectively. (S)-cEt-BNA (S)-constrained ethyl bicyclic nucleic acid, PMO phosphorodiamidate morpholino oligomer
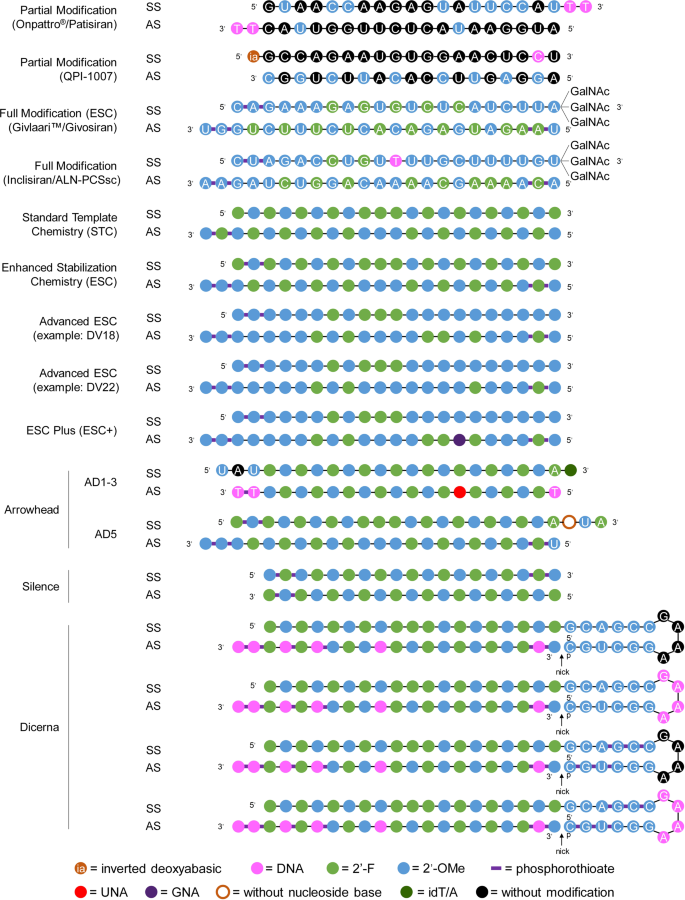
Representative designs for the chemical modification of siRNA. The sequences and modification details for ONPATTRO®, QPI-1007, GIVLAARI™ and inclisiran are included. The representative siRNA modification patterns developed by Alnylam (STC, ESC, advanced ESC and ESC+) and arrowhead (AD1-3 and AD5) are shown. Dicerna developed four GalNAc moieties that can be positioned at the unpaired G–A–A–A nucleotides of the DsiRNA structure. 2′-OMe 2′-methoxy, 2′-F 2′-fluoro, GNA glycol nucleic acid, UNA unlocked nucleic acid, SS sense strand, AS antisense strand
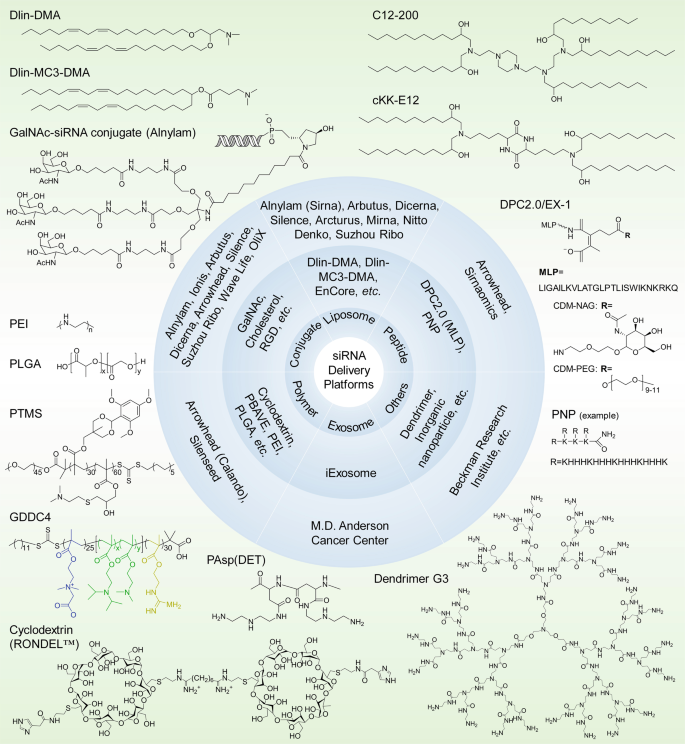
siRNA delivery platforms that have been evaluated preclinically and clinically. Varieties of lipids or lipidoids, siRNA conjugates, peptides, polymers, exosomes, dendrimers, etc. have been explored and employed for siRNA therapeutic development by biotech companies or institutes. The chemical structures of the key component(s) of the discussed delivery platforms, including Dlin-DMA, Dlin-MC3-DMA, C12-200, cKK-E12, GalNAc–siRNA conjugates, MLP-based DPC2.0 (EX-1), PNP, PEI, PLGA-based LODER, PTMS, GDDC4, PAsp(DET), cyclodextrin-based RONDEL™ and dendrimer generation 3 are shown. DLin-DMA (1,2-dilinoleyloxy-3-dimethylaminopropane), DLin-MC3-DMA (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate, DPC Dynamic PolyConjugates, MLP membrane-lytic peptide, CDM carboxylated dimethyl maleic acid, PEG polyethylene glycol, NAG N-acetylgalactosamine, PNP polypeptide nanoparticle, PEI poly(ethyleneimine), LODER LOcal Drug EluteR, PLGA poly(lactic-co-glycolic) acid, PTMS PEG-PTTMA-P(GMA-S-DMA) poly(ethylene glycol)-co-poly[(2,4,6-trimethoxybenzylidene-1,1,1-tris(hydroxymethyl))] ethane methacrylate-co-poly(dimethylamino glycidyl methacrylate), GDDC4 PG-P(DPAx-co-DMAEMAy)-PCB, where PG is guanidinated poly(aminoethyl methacrylate) PCB is poly(carboxybetaine) and P(DPAx-co-DMAEMAy) is poly(dimethylaminoethyl methacrylate-co-diisopropylethyl methacrylate), PEG-PAsp(DET) polyethylene glycol-b-poly(N′-(N-(2-aminoethyl)-2-aminoethyl) aspartamide), PBAVE polymer composed of butyl and amino vinyl ether, RONDEL™ RNAi/oligonucleotide nanoparticle delivery
| Vutrisiran Sodium
Vutrisiran Sodium is a sodium salt of an siRNA derivative targeting transthyretin (TTR) covalently linked to a triantennary GalNAc3 complex at the 3’ end of the sense strand. The siRNA moiety is composed of a duplex oligonucleotide of sense strand consisting of chemically modified 21 nucleotide residues and antisense strand consisting of chemically modified 23 nucleotide residues each. C530H672F9N171Na43O323P43S6 : 17289.77 |
REF
Nucleic Acids Research (2019), 47(7), 3306-3320.
Drug Metabolism & Disposition (2019), 47(10), 1183-1201.
PATENT
WO 2020128816
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020128816
The present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof a combination of a benzoxazole derivative transthyretin stabilizer or a pharmaceutically acceptable salt or prodrug thereof and an additional therapeutic agent for the treatment of transthyretin amyloidosis. Particularly, the present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent for the treatment of transthyretin amyloidosis.
The present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof a combination of a benzoxazole derivative transthyretin stabilizer or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent. Particularly, the present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent. The compositions and methods of the invention are useful in stabilizing transthyretin, inhibiting transthyretin misfolding, proteolysis, and treating amyloid diseases associated thereto.
Transthyretin (TTR) is a 55 kDa homotetrameric protein present in serum and cerebral spinal fluid and which functions as a transporter of L-thyroxine (T4) and holo-retinol binding protein (RBP). TTR has been found to be an amyloidogenic protein that, under certain conditions, can be transformed into fibrils and other aggregates which can lead to disease pathology such as polyneuropathy or cardiomyopathy in humans.
US Patent Nos. 7,214,695; 7,214,696; 7,560,488; 8, 168.683; and 8,653,119 each of which is incorporated herein by reference, discloses benzoxazole derivatives which act as transthyretin stabilizers and are of the formula
or a pharmaceutically acceptable salt thereof; wherein Ar is 3,5-difluorophenyl, 2,6-difluorophenyl, 3,5-dichlorophenyl, 2,6-dichlorophenyl, 2-(trifluoromethyl)phenyl or 3-(trifluoromethyl)phenyl. Particularly, 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis) of the formula
is disclosed therein. Tafamidis is an orally active transthyretin stabilizer that inhibits tetramer dissociation and proteolysis that has been approved in certain jurisdictions for the treatment of transthyretin polyneuropathy (TTR-PN) and is currently in development for the treatment of transthyretin cardiomyopathy (TTR-CM). US Patent No. 9,249, 112, also incorporated herein by reference, discloses polymorphic forms of the meglumine salt of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis meglumine). US Patent No. 9,770,441 discloses polymorphic forms of the free acid of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis), and is also incorporated by reference herein.
Summary of the Invention
The present invention provides pharmaceutical compositions and methods comprising the compound 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent. Particular embodiments of this invention are pharmaceutical compositions and methods comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agents selected from the group consisting of agents that lower plasma levels of TTR such as an antisense therapy, TTR gene editing therapy, transcriptional modulators, translational modulators, TTR protein degraders and antibodies that bind and reduce TTR levels; amyloid reduction therapies such as anti amyloid antibodies (either TTR selective or general), stimulators of amyloid clearance, fibril disruptors and therapies that inhibit amyloid nucleation; other TTR stabilizers; and TTR modulators such as therapeutics which inhibit TTR cleavage. Particularly, the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and
methods comprising a polymorphic form of tafamidis free acid or a polymorphic form of tafamidis meglumine salt with one or more additional therapeutic agents.
The present invention also provides a method of treating or preventing transthyretin amyloidosis in a patient, the method comprising administering to a patient in need thereof a therapeutically or prophylactically effective amount of 2-(3,5-dichlorophenyl)-1,3-benzoxazole- 6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agents.
A particular embodiment of the present method of treatment is the method comprising a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent are administered orally. Additional embodiments of this invention are methods of treatment as described above wherein the 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent are administered parenterally (intravenously or subcutaneously). Further embodiments of this invention are methods of treatment wherein the 2-(3,5-dichlorophenyl)-1, 3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally and the one or more additional therapeutic agent is administered either orally or parenterally. Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR 5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR 5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR
cardiomyopathy, the method comprising administering to a patient in need thereof a therapeutically effective amount of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agents.
Brief Description of the Drawings
REF
Biochemical Pharmacology (Amsterdam, Netherlands) (2021), 189, 114432.
PATENT
WO 2021041884
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021041884
Exemplary RNAi agents that reduce the expression of TTR include patisiran and vutrisiran.
The ter s “antisense polynucleotide agent”, “antisense oligonucleotide”, “antisense compound”, and “antisense agent” as used interchangeably herein, refer to an agent comprising a single-stranded oligonucleotide that specifically binds to the target nucleic acid molecules via hydrogen bonding (e.g., Watson-Crick, Hoogsteen, or reversed Hoogsteen hydrogen bonding) and inhibits the expression of the targeted nucleic acid by an antisense mechanism of action, e.g., by RNase H. In some embodiments, an antisense agent is a nucleic acid therapeutic that acts by reducing the expression of a target gene, thereby reducing the expression of the polypeptide encoded by the target gene. Exemplary antisense agents that reduce the expression of TTR include inotersen and Ionis 682884/ ION-TTR-LRx (see, e.g., WO2014179627 which is incorporated by reference in its entirety). Further antisense agents that reduce the expression of TTR are provided, for example in WO2011139917 and WO2014179627, each of which is incorporated by reference in its entirety.
REF
Clinical Pharmacology & Therapeutics (Hoboken, NJ, United States) (2021), 109(2), 372-382
Annals of Plastic Surgery (2021), 86(2S_Suppl_1), S23-S29.
Journal of Cardiovascular Pharmacology (2021), 77(5), 544-548.
Annals of Pharmacotherapy (2021), 55(12), 1502-1514.
Kidney International (2022), 101(2), 208-211
//////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
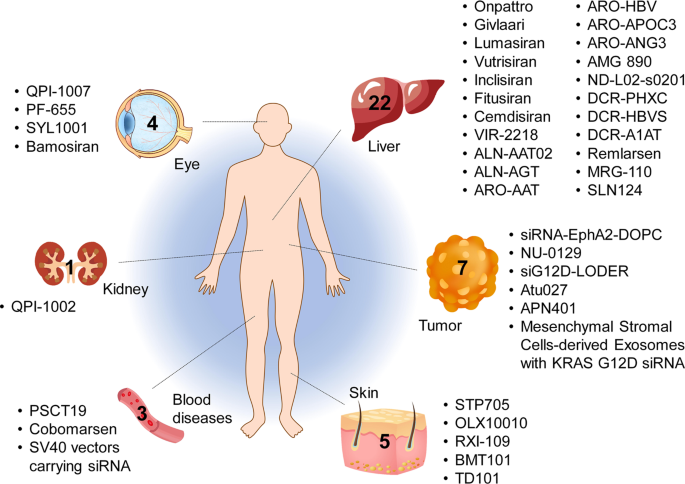
Tissues targeted by siRNA and miRNA therapeutics currently being investigated at the clinical stage. The corresponding therapeutic names are shown beside the tissues
CLIP
https://www.alnylam.com/sites/default/files/pdfs/Vutrisiran-Fact-Sheet.pdf
Vutrisiran An Investigational RNAi Therapeutic for ATTR Amyloidosis Vutrisiran has not been approved by the U.S. Food and Drug Administration, European Medicines Agency, or any other regulatory authority and no conclusions can or should be drawn regarding the safety or effectiveness of this investigational therapeutic. Overview • Vutrisiran is an investigational RNAi therapeutic in development for the treatment of transthyretin-mediated (ATTR) amyloidosis, which encompasses both hereditary ATTR (hATTR) amyloidosis and wild-type ATTR (wtATTR) amyloidosis.1, 2 • Vutrisiran inhibits the production of disease-causing transthyretin (TTR) protein by the liver, leading to a reduction in the level of TTR in the blood.1, 2 • Vutrisiran is administered subcutaneously (under the skin) and utilizes one of Alnylam’s delivery platforms known as the Enhanced Stabilization Chemistry (ESC)-GalNAc-conjugate delivery platform.1, 2 • Vutrisiran is administered every three months.2 • Vutrisiran is under review by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the Brazilian Health Regulatory Agency (ANVISA). Vutrisiran has been granted Orphan Drug Designation in the U.S. and the European Union (EU) for the treatment of ATTR amyloidosis. Vutrisiran has also been granted a Fast Track designation in the U.S. for the treatment of the polyneuropathy of hATTR amyloidosis in adults. In the U.S. vutrisiran has received an action date under the Prescription Drug User Fee Act (PDUFA) of April 14, 2022. The Company received orphan drug designation in Japan. Alnylam has global commercial rights to vutrisiran, assuming regulatory approvals. Clinical Development • A Phase 1 clinical study of vutrisiran was conducted in 80 healthy volunteers (60 received vutrisiran and 20 received placebo). Vutrisiran demonstrated an acceptable safety profile and a single dose reduced serum TTR for a period of at least 90 days.2 • The safety and efficacy of vutrisiran are being evaluated in the HELIOS Phase 3 clinical program, currently consisting of two clinical trials: HELIOS-A and HELIOS-B. • HELIOS-A is a randomized, open-label, global multi-center Phase 3 study of 164 adult patients with hATTR amyloidosis with polyneuropathy.1 • The primary endpoint of HELIOS-A is change from baseline in the modified Neuropathy Impairment Score +7 (mNIS+7) at 9 months. • Secondary endpoints at 9 months include the Norfolk Quality of Life-Diabetic Neuropathy (Norfolk QoL-DN) Total Score and the 10-Meter Walk Test (10-MWT). • The 9-month endpoints will be analyzed at 18 months with the addition of other secondary endpoints. • HELIOS-B is a randomized, double-blind, placebo-controlled Phase 3 study of 655 adult patients with ATTR amyloidosis with cardiomyopathy (including both hATTR and wtATTR amyloidosis).3 • The primary endpoint will evaluate the efficacy of vutrisiran versus placebo for the composite outcome of all-cause mortality and recurrent cardiovascular (CV) events (CV hospitalizations and urgent heart failure (HF) visits) at 30-36 months. • Secondary endpoints include the change from baseline in the 6-minute walk test (6-MWT), health status measured using the Kansas City Cardiomyopathy Questionnaire Overall Summary (KCCQ-OS), echocardiographic assessments of mean left ventricular wall thickness and global longitudinal strain, the N-terminal prohormone B-type natriuretic peptide (NT-proBNP) as a cardiac biomarker, and all-cause mortality, rate of recurrent CV events, and composite of all-cause mortality and recurrent all-cause hospitalizations and urgent HF visits at month 30 or 30-36 months. Page 2 © 2021 Alnylam Pharmaceuticals, Inc. All rights reserved. TTRsc02-USA-00012 v4 About ATTR Amyloidosis • ATTR amyloidosis is a rare, underdiagnosed, rapidly progressive, debilitating, and fatal disease caused by misfolded TTR that accumulates as amyloid fibrils in multiple tissues including the nerves, heart, and GI tract. There are two types of ATTR amyloidosis: hATTR amyloidosis and wtATTR amyloidosis.4,5,6 • hATTR amyloidosis is an inherited condition that is caused by variants (i.e., mutations) in the transthyretin (TTR) gene.5,7,8 TTR protein is produced primarily in the liver and is normally a carrier of vitamin A.9 The variant results in misfolded TTR proteins that accumulate as amyloid deposits in multiple tissues, including the nerves, heart and gastrointestinal (GI) tract.5, 6, 7 It is a multisystem disease that can include sensory and motor, autonomic, and cardiac symptoms. The condition can have a debilitating impact on a patient’s life and may lead to premature death with a median survival of 4.7 years following diagnosis.8,10 It is estimated that there are approximately 50,000 patients with hATTR amyloidosis worldwide.11 • wtATTR amyloidosis is a non-hereditary condition that occurs when misfolded wild-type TTR accumulates as amyloid deposits in multiple organs. It predominantly manifests as cardiac symptoms, but other systems are also involved, and commonly leads to heart failure and mortality within 2.5 to 5.5 years.12,13,14,15,16,17,18,19 wtATTR amyloidosis affects an estimated 200,000-300,000 people worldwide.20 • Alnylam is committed to developing multiple treatment options for people who are living with ATTR amyloidosis to help manage the debilitating and progressive nature of the disease. For more information about vutrisiran, please contact media@alnylam.com. For more information on HELIOS-A (NCT03759379) and HELIOS-B (NCT04153149) please visit www.clinicaltrials.gov or contact media@alnylam.com. Current information as of November 2021
CLIP
Alnylam announces extension of review period for new drug vutrisiran to treat ATTR amyloidosis
Alnylam announces 3-month extension of review period for new drug application for vutrisiran to treat ATTR amyloidosis.
Alnylam Pharmaceuticals, Inc., a RNAi therapeutics company, announced that the FDA has extended the review timeline of the New Drug Application (NDA) for vutrisiran, an investigational RNAi therapeutic in development for the treatment of transthyretin-mediated (ATTR) amyloidosis, to allow for the review of newly added information related to the new secondary packaging and labelling facility.
Alnylam recently learned that the original third-party secondary packaging and labelling facility the Company planned to use for the vutrisiran launch was recently inspected and the inspection requires classification for the FDA to take action on the vutrisiran NDA. The inspection observations were not directly related to vutrisiran. In order to minimize delays to approval, Alnylam has identified a new facility to pack and label vutrisiran and submitted an amendment to the NDA for review by the FDA. The updated Prescription Drug User Fee Act (PDUFA) goal date to allow for this review is July 14, 2022. No additional clinical data have been requested by the FDA.
////////////Vutrisiran sodium, APPROVALS 2022, FDA 2022, FDA APPROVED, AMVUTTRA, 2022/6/13, ブトリシランナトリウム , ALN 65492, Votrisiran, siRNA