tert-butyl 4-(2-methoxy-2-oxoethylidene)piperidine-l -carboxylate. Sodium hydride in mineral oil (60%, 7.92 g, 198.02 mmoles) was washed with hexanes then suspended in dimethylformamide (220 mL). The mixture was cooled to 0°C. Trimethyl phosphonoacetate (29.0 mL, 189.82 mmoles) was added dropwise to the stirred reaction mixture. After 20 min at 0°C, a solution of A-/c/7-butoxycarbonyl-4-pi peri done (30.41 g, 152.62 mmoles) in dimethylformamide (80 mL) was added to the mixture dropwise. The reaction was stirred at room temperature for 3 h and then diluted with diethyl ether (650 mL). The mixture was washed once with water and the aqueous layer was extracted once with diethyl ether. The combined organic layers were washed 4 times with water and the aqueous phase was discarded. The organic phase was washed with brine and dried over magnesium sulfate, filtered, and concentrated to dryness. The title compound was obtained as a white solid in 92% yield.
1 H- NMR (300 MHz, CDCh): 5 = 5.68 (s, 1 H), 3.66 (s, 3 H), 3.40-3.51 (m, 4 H), 2.90 (t, J= 5.49, 2 H), 2.25 (t, J= 5.49, 2 H), 1.44 (s, 9 H).
ed-butyl 4-(2-methoxy-2-oxoethyl)piperidine-l -carboxylate. A solution of tert-butyl 4- (2-methoxy-2-oxoethylidene)piperidine-l -carboxylate (35.71 g, 140 mmoles) in a mixture of 1 : 1 ethyl acetate/methanol (220 mL) was carefully treated with 50% wet 10% palladium on carbon (3.3 g). The reaction vessel was charged with 55 psi of hydrogen gas and the mixture was shaken on a Parr apparatus at room temperature for 16 h. The reaction mixture was then filtered to remove the catalyst and the filtrate concentrated in vacuo. The title compound was obtained as a clear colorless oil in 97% yield. ‘H-NMR (300 MHz, CDCh): 5 = 4.04 (d, J= 10.25, 2 H), 3.64 (s, 3 H), 2.68 (t, J= 12.44, 2 H), 2.21 (d, J= 6.95, 2 H), 1.98-1.77 (m, 1 H), 1.64 (d, J= 13.54, 2 H), 1.41 (s, 9 H), 1.25-0.99 (m, 2 H).
4-[2-Hydroxy-l-methoxycarbonyl-2-(2-nitro-phenyl)-ethyl]-piperidine-l-carboxylic acid tert-butyl ester. A A-diisopropylamine (4.40 mL, 31.3 mmoles) was dissolved in tetrahydrofuran (50 mL). The mixture was cooled to -78°C. Butyllithium (2.5 M in hexanes, 12.4 mL, 31 mmoles) was added dropwise to the stirred solution. After stirring at -78°C for 30 min, a solution of tert-butyl 4-(2-methoxy-2-oxoethyl)piperidine-l -carboxylate (6.65 g, 25.8 mmoles) in tetrahydrofuran (15 mL) was added dropwise to the mixture. Stirring was continued at -78°C for 1 h. A solution of 2-nitrobenzaldehyde (3.90 g, 25.8 mmoles) in tetrahydrofuran (20 mL) was then added to the mixture dropwise, and then stirring was continued at -78°C for a further 2.5 h. The reaction was quenched with cold aqueous ammonium chloride and then diluted with water. The mixture was extracted twice with ethyl acetate and the aqueous phase was discarded. The material was dried (magnesium sulfate) filtered, and concentrated to dryness. Silica gel chromatography afforded the desired product in 94% yield as light yellow foam. MS m/e (M- C4H8+H)+= 353.1.
4-(4-Hydroxy-2-oxo-l , 2, 3, 4-tetrahydro-quinolin-3-yl)-piperidine-l -carboxylic acid tertbutyl ester. In a 3 neck flask fitted with a nitrogen inlet, thermometer, and a mechanical stirrer, 4-[2-hydroxy-l -methoxy carbonyl-2-(2-nitro-phenyl)-ethyl]-piperidine-l -carboxylic acid tertbutyl ester (9.93 g, 24.3 mmoles) was dissolved in acetic acid (1.75 moles, 100 mL). Iron powder (8.90 g, 159 mmoles) was added to the vessel with stirring. The stirred mixture was slowly heated to 80°C for 30 min and then cooled to room temperature. It was then diluted with ethyl acetate and filtered through a pad of celite. Solids were washed with 20% methanol/ethyl acetate, and then with methanol. The filtrate was concentrated and the residue partitioned between ethyl acetate and aqueous sodium bicarbonate. The layers were separated. The resulting aqueous phase was extracted twice with ethyl acetate. The organic layers were combined. The mixture was washed twice with water and the aqueous phase was discarded. The material was dried (magnesium sulfate) filtered, and concentrated to dryness. Silica gel chromatography afforded the title compound as light yellow foam in 77% yield. MS m/e (M-H)’ = 345.1.

3-(Piperidin-4-yl)quinolin-2(lH) hydrochloride . A stirred solution of 4-(4-hydroxy-2- oxo-l,2,3,4-tetrahydro-quinolin-3-yl)-piperidine-l-carboxylic acid tert-butyl ester (5.60 g, 16.2 mmoles) in ethyl acetate (70 mL) was treated with HC1 in dioxane (4N, 40 mmoles, 10 mL). The mixture was stirred at room temperature for 45 min. More HC1 in dioxane (4N, 120 mmoles, 30 mL) was then added and stirring was continued at room temperature for 16 h. The resulting solid was collected by filtration and washed with ethyl acetate. It was then suspended in 5% water-isopropanol (100 mL) and the mixture was warmed to reflux and stirred for 20 min. The mixture was cooled to room temperature and stirred at room temperature for 16 h. The solid was collected by filtration, washed with isopropanol, and dried under high vacuum. The title compound was obtained as white solid in 75% yield. ‘H-NMR (DMSO-de) 5 11.85 (s, 1 H), 9.02 (bs, 1 H), 8.88 (bs, 1 H), 7.70 (t, J= 3.81 Hz, 2 H), 7.53 – 7.30 (d, J= 8.24 Hz, 1 H), 7.17 (t, J= 7.48 Hz, 2 H), 3.36 (d, J= 12.51 Hz, 2 H), 3.10 – 2.94 (m, 3 H), 2.01 (d, J= 13.43 Hz, 2 H), 1.87 – 1.73 (m, 2 H); MS m/e (M+H)
+ = 229.0.
4-Iodo-2,6-dimethylbenzenamine hydrochloride . To a suspension of sodium bicarbonate (126 g, 1.5 moles) and 2,6-dimethylaniline (61.5 mL, 500 mmoles) in methanol (700 mL) was added iodine monochloride (1.0 M in dichloromethane, 550 mL, 550 mmoles) at room temperature over 1 h. After addition was complete, stirring was continued for 3 h. The reaction was filtered to remove excess sodium bicarbonate and the solvent removed in vacuo. The residue was re-dissolved in diethyl ether (1.5 L) and treated with hydrochloric acid (2M in ether, 375 mL, 750 mmoles). The resulting suspension was stored in the freezer (-15°C) overnight. The solid was filtered and washed with diethyl ether until it became colorless, to give 126.5 g (89%) as a grey-green powder. ‘H-NMR (DMSO-de) 5 2.33 (s, 6 H), 7.48 (s, 2 H), 9.05 (bs, 3 H);
13C-NMR (DMSO-de) 5 17.4, 91.5, 133.1, 131.2, 136.9.
Methyl 2 -(benzyloxy carbonyl) acrylate . To a flame dried three-neck round bottom flask, fitted with a mechanical stirrer, was added (S)-methyl 2-(benzyloxycarbonyl)-3- hydroxypropanoate (129 g, 509 mmoles), anhydrous dichloromethane (2 L), and methanesulfonyl chloride (49.3 mL, 636 mmoles). The mixture was cooled to -15°C, and treated with tri ethylamine (213 mL, 1527 mmoles), dropwise, to ensure the temperature of the reaction mixture did not exceed 0°C. The addition of the first equivalent of triethylamine was exothermic. After addition of tri ethylamine, the mixture was stirred at 0°C for 30 min. The cooling bath was removed and the mixture stirred at room temperature for 1.5 h. The reaction was quenched by addition of methanol (21 mL). The mixture was washed with 0.5% aqueous potassium bisulfate until the washings were pH 5, then saturated sodium bicarbonate, and brine, dried over sodium sulfate, and concentrated. Flash chromatography (silica gel, 1 :9 ethyl acetate/hexanes) gave I l l g (92%) as a viscous colorless oil, which crystallized upon standing. ’H-NMR (DMSO-de) 5 3.71 (s, 3 H), 5.10 (s, 2 H), 5.60 (s, 1 H), 5.76 (s, 1 H), 7.39-7.35 (m, 5 H), 8.96 (s, 1 H);
13C-NMR (DMSO-de) 5 52.3, 65.9, 127.8, 128.1, 128.3, 128.8, 133.3, 136.3, 153.5, 163.7.
(Z)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl) acrylate. A 2 L round bottom flask was charged 4-iodo-2,6-dimethylbenzenamine hydrochloride salt (55 g, 194 mmoles), methyl 2-(benzyloxycarbonyl)acrylate (59.2 g, 252 mmoles), tetrabutylammonium chloride (59.2 g, 213 mmoles), palladium (II) acetate (4.34 g, 19.4 mmoles), and tetrahydrofuran (1.2 L, degassed by a flow of nitrogen for 30 min). The mixture was stirred so that a suspension was formed and then degassed by a flow of nitrogen for 30 min. Triethylamine (110 mL, 789 mmoles) was added and the resulting mixture was heated at reflux for 3 h. After cooling to room temperature, the reaction mixture was filtered through a pad of celite, washed with tetrahydrofuran (2 x 100 mL), and concentrated. The residue was dissolved in di chloromethane, washed with water (3X) and brine (2X), dried over sodium sulfate, and concentrated. Flash chromatography (silica gel, using 1 :9 ethyl acetate/dichloromethane) gave a tan solid. The solid was recrystallized from warm methanol (210 mL) and water (100 mL). The mixture was held at room temperature overnight, then at 0°C for 2 h, and finally at -15°C for 2 h. The resulting solid was filtered, washed with ice cold 1 : 1 methanol/water, and dried under high vacuum overnight to give 44.7 g (65%) as a light tan solid which was a mixture of ZZE isomers (73 :27). ’H-NMR (DMSO-de) 5, 2.05 (s, 6 H), 3.61 (s, 0.8 H), 3.68 (s, 2.2 H), 5.00 (s, 0.54 H), 5.13 (s, 1.46 H), 5.24 (s, 2 H), 7.40-7.21 (m, 8 H), 8.51 (s, 0.27 H), 8.79 (s, 0.73 H);
13C-NMR (DMSO-de) 5 17.8, 51.7, 65.3, 119.4, 120.0, 120.3, 127.3, 127.7, 128.3, 130.9, 135.8, 137.2, 146.9, 154.7, 166.0.
(R)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl)propanoate. A flame- dried 2 L Parr hydrogenation bottle was charged with (Z)-methyl 3-(4-amino-3,5- dimethylphenyl)-2-(benzyloxycarbonyl)acrylate (84.5 g, 239 mmoles), di chloromethane (300 mL), and methanol (300 mL). The bottle was swirled so that a light brown suspension was formed. The mixture was degassed using a flow of nitrogen for 30 min. To this was quickly added (-)-l,2-bis((2A,5A)-2,5-diethylphospholano)-bezene(cyclooctadiene) rhodium (I) tetrafluoroborate ([(2A,5A)-Et-DuPhosRh]BF4) (2.11 g, 3.20 mmoles). The bottle was immediately attached to a Parr Hydrogenator. After 5 cycles of hydrogen (60 psi) and vacuum, the bottle was pressurized to 65 psi and the suspension was agitated at room temperature for 16 h. The reaction had become homogeneous. The reaction mixture was concentrated, and the resulting residue purified by flash chromatography (silica gel, 1 :9 ethyl acetate/dichloromethane) to give 82.9 g (98%). ‘H-NMR (DMSO-de) 5 2.04 (s, 6 H), 2.65 (dd, J= 13.4, 9.8 Hz, 1H), 2.82 (dd, J= 13.7, 5.2 Hz, 1 H), 3.62 (s, 3 H), 4.15-4.10 (m, 1H), 4.41 (s, 2 H), 5.00 (s, 2 H), 6.68 (s, 2 H), 7.37-7.28 (m, 5 H), 7.70 (d, J= 7.9 Hz, 1 H);
13C-NMR (DMSO-de) 5 17.7, 35.9, 51.7, 56.1, 65.3, 120.4, 124.0, 127.5, 127.7, 128.2, 128.3, 136.9, 142.6, 155.9, 172.5.
(R)-Methyl 2-(benzyloxycarbonyl)-3-(7-methyl-lH-indazol-5-yl)propanoate. (R)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl)propanoate (50.0 g, 140 mmoles) was weighed into a flame-dried 5 L three neck round bottom flask, followed by the addition of toluene (2.4 L) and glacial acetic acid (120 mL, 2.1 moles). The mixture was mechanically stirred to form a clear solution, and then potassium acetate (103 g, 1.05 moles) was added. To the resulting white suspension, z.w-amyl nitrite (20.7 mL, 154 mmoles) was added dropwise at room temperature, and the resulting mixture was stirred at room temperature for 16 h. Saturated sodium bicarbonate (I L) was added, followed by the careful addition of solid sodium bicarbonate to neutralize the acetic acid. The mixture was extracted with a mixture of di chloromethane (2 L) and brine (1.5 L). After separation, the aqueous layer was extracted with di chloromethane (500 mL). The combined organic layers were dried over anhydrous sodium sulfate and filtered. Solvents were removed to afford a tan solid, which was washed with hexanes (2 L) and toluene (150 mL). The solid was recrystallized from hot acetone (260 mL) and hexanes (700 mL). The slightly cloudy mixture was allowed to cool to room temperature slowly, then to 0°C for 1.5 h, and finally to -15°C for 1.5 h. The resulting solid was filtered and washed with ice-cold acetone/hexanes (1 : 1, 200 mL) to afford 39.1 g (76% yield). Analytical HPLC showed >98% UV purity. The enantiomeric excess (ee) was determined to be 99.8% (conditions: Chiralpak AD column, 4.6 x 250 mm, 10 pm; A = ethanol, B = 0.05% diethylamine/heptane; 85%B @1.0 mL/min. for 55 min. The retention times for R was 44.6 min and for S was 28.8 min). ‘H-NMR (DMSO-de) 5 2.48 (s, 3 H), 2.93 (dd, J= 13.4, 10.7 Hz, 1H), 3.10 (dd, J= 13.7, 4.9 Hz, 1H), 3.63 (s, 3H), 4.32-4.27 (m, 1 H), 4.97 (s, 2 H), 7.03 (s, 1 H), 7.24-7.22 (m, 2 H), 7.29 -7.27 (m, 3 H), 7.41 (s, 1 H), 7.83 (d, J= 8.2 Hz, 1H), 7.99 (s, 1H), 13.1 (s, 1 H);
13C-NMR (DMSO-de) 5 16.7, 36.5, 51.8, 56.0, 65.3, 117.6, 119.6, 122.7, 127.2, 127.4, 127.6, 128.2, 129.3, 133.4, 136.8, 139.2, 155.9, 172.4. Mass spec.: 368.16 (MH)
+.
(R)-Methyl 2-amino-3-(7-methyl-lH-indazol-5-yl)propanoate. A Parr hydrogenation bottle was charged with (R)-methyl 2-(benzyloxycarbonyl)-3-(7-methyl-lH-indazol-5- yl)propanoate (11.0 g, 29.9 mmoles) and methanol (75 mL). The suspension was purged with nitrogen and treated with palladium (10% on charcoal, 700 mg). The bottle was shaken under hydrogen (15 psi) overnight. The mixture was filtered through a pad of celite to remove the catalyst. Concentration of the eluent gave 7.7 g (quant.) as an oil which was used without further purification.
XH-NMR (CD
3OD) 5 2.54 (s, 3 H), 2.98 (dd, J= 13.5, 7.0 Hz, 1 H), 3.09 (dd, J= 13.5, 5.9 Hz, 1 H), 3.68 (s, 3 H), 3.75 (dd, J= 7.0, 6.2 Hz, 1 H), 7.01 (s, 1 H), 7.39 (s, 1 H), 7.98 (s, 1 H). Mass spec.: 232.34 (M-H)’.
(R)-methyl 3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3- yl)piperidine-l-carboxamido)propanoate. To a solution of (R)-methyl 2-amino-3-(7-methyl-lH- indazol-5-yl)propanoate hydrochloride (7.26 g, 27.0 mmoles) in dimethylformamide (50 mL) at room temperature was added N, A’-disuccinimidyl carbonate (7.60 g, 29.7 mmoles) followed by triethylamine (11.29 mL, 81 mmoles). The resulting mixture was stirred for 30 min and treated with 3-(piperidin-4-yl)quinolin-2(lH)-one (6.77 g, 29.9 mmoles) in portions. The reaction was allowed to stir for 24 h. The mixture was concentrated, dissolved in ethyl acetate, and washed sequentially with water, brine, and 0.5 N HC1 (2X). The organic phase was dried over magnesium sulfate, filtered, and concentrated. The resulting residue was purified by flash chromatography (silica gel, 20: 1 ethyl acetate/methanol) to give 11.9 g (78%).
1 H-NMR (CD3OD) 5 13.0 (s, 1 H), 11.8 (s, 1 H), 7.98 (s, 1 H), 7.63 (d, J= 7.6 Hz, 1 H), 7.57 (s, 1 H), 7.45 – 7.41 (m, 2 H), 7.27 (d, J= 8.2Hz, 1 H), 7.16 (t, J= 7.9 Hz, 1 H), 7.03 (s, 1 H), 6.85 (d, J= 7.9 Hz, 1 H), 4.31 – 4.26 (m, 1 H), 4.10 – 4.08 (m, 2 H), 3.60 (s, 3 H), 3.07 – 3.01 (m, 2 H), 2.93 – 2.88 (m, 1 H), 2.77 – 2.67 (m, 2 H), 2.48 (s, 3 H), 1.78 – 1.72 (m, 2 H), 1.34 – 1.26 (m, 2 H). Mass spec.: 488.52 (MH)
+.
(R)-3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3-yl)piperidine-l- carboxamido)propanoic acid. A solution of (R)-methyl 3-(7-methyl-lH-indazol-5-yl)-2-(4-(2- oxo-1, 2-dihydroquinolin-3-yl)piperidine-l-carboxamido)propanoate_(5.50 g, 11.3 mmoles) in tetrahydrofuran (50 mL) and methanol (10 mL) was cooled to 0°C. To this was added a cold (0°C) solution of lithium hydroxide monohydrate (0.95 g, 22.6 mmoles) in water (20 mL), dropwise over 15 min. The reaction was stirred at room temperature for additional 3 h. The mixture was concentrated to remove the organic solvents. The resulting residue was dissolved in a minimum amount of water, cooled to 0°C, and treated with cold (0°C) IN HC1 until pH 2 was attained. The resulting solid was collected by filtration, washed with cold water and ether, and then dried overnight under high vacuum to give 5.0 g (94%) as a white solid. ’H-NMR (DMSO- d6) 5 13.05 (bs, 1 H), 11.77 (s, 1 H), 7.98 (s, 1 H), 7.62 (d, J= 8.0 Hz, 1 H), 7.55 (s, 1 H), 7.44 (d, J= 8.2Hz, 1 H), 7.42 (s, 1 H), 7.27 (d, J= 8.2 Hz, 1 H), 7.16 (t, J= 7.6 Hz, 1 H), 7.05 (s, 1 H), 6.65 (d, J= 7.9 Hz, 1 H), 4.27 – 4.22 (m, 1 H), 4.10 – 4.07 (m, 2 H), 3.12 – 3.07 (m, 1 H), 3.03 – 2.99 (m, 1 H), 2.93 – 2.88 (m, 1 H), 2.77 – 2.66 (m, 2 H), 2.47 (s, 3 H), 1.77 – 1.74 (m, 2 H), 1.34 – 1.27 (m, 2 H). Mass spec.: 474.30 (MH)+.
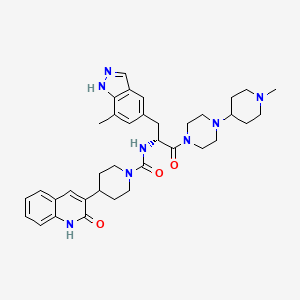
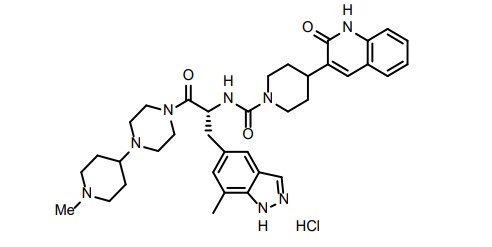
(R)-N-(3-(7-methyl-lH-indazol-5-yl)-l-(4-(l-methylpiperidin-4-yl)piperazin-l-yl)-l- oxopropan-2-yl)-4-(2-oxo-l,2-dihydroquinolin-3-yl)piperidine-l-carboxamide (I). A flask was charged with (R)-3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3- yl)piperidine-l-carboxamido)propanoic acid (2.9 g, 6.11 mmoles), triethylamine (3.00 mL, 21.5 mmoles), l-(l-methylpiperidin-4-yl)piperazine (1.23 g, 6.72 mmoles), and dimethylformamide (10 mL). The resulting solution was treated with 2-(lH-benzotriazole-l-yl)-l, 1,3,3- tetramethyluronium tetrafluoroborate (2.26 g, 7.03 mmoles) in portions. The reaction was allowed to stir at room temperature overnight. The mixture was concentrated under vacuum to remove dimethylformamide. The crude product was dissolved in 7% methanol in di chloromethane and purified by flash chromatography using 7% methanol in di chloromethane containing 2% of aqueous ammonium hydroxide as eluent. The pure fractions were collected and solvent was removed under vacuum. The desired product was crystallized from hot acetone to give the compound having Formula I in 77% yield. Analytical HPLC showed 99.0 % UV purity at 230 nm. The enantiomeric excess (ee) was determined to be >99.9% (conditions: Chiralpak AD column, 4.6 x 250 mm, 10 pm; eluent: 70% (0.05% diethylamine)/heptane/30%ethanol; @1.0 mL/min. for 45 min. The retention times were 18.7 min for R and 28.1 min for S). ‘H-NMR (500 MHz, DMSO-de) 5 ppm 13.01 (s, 1 H), 11.76 (s, 1 H), 7.96 (s, 1 H), 7.62 (d, J= 7.10 Hz, 1 H), 7.60 (s, 1 H), 7.42 (m, 1 H), 7.36 (s, 1 H), 7.26 (d, J = 8.25 Hz, 1 H), 7.14 (m, 1 H), 7.00 (s, 1 H), 6.69 (d, J= 8.25 Hz, 1 H), 4.78 (q, J= 7.79 Hz, 1 H), 4.14 (d, J= 12.37 Hz, 2 H), 3.54 (dd, J= 9.16, 4.58 Hz, 1 H), 3.24 (m, 1 H), 3.11 (m, 1 H), 2.97 (m, 1 H), 2.89 (m, 2 H), 2.69 (m, 4 H), 2.32 (m, 1 H), 2.21 (m, 1 H), 2.07 (m, 4 H), 1.95 (t, J= 8.25 Hz, 1 H), 1.87 (m, J= 11.28, 11.28, 3.55, 3.44 Hz, 1 H), 1.76 (t, J= 12.03 Hz, 2 H), 1.68 (t, J= 11.11 Hz, 2 H), 1.53 (t, J= 8.25 Hz, 1 H), 1.32 (m, 4 H), 1.16 (m, 2 H); 13C-NMR (DMSO-de) 5 16.80, 27.30, 30.51, 30.51, 30.67, 35.50, 38.04, 41.74, 44.00, 44.16, 45.35, 45.78, 48.14, 48.39, 51.45, 54.76, 54.76, 60.61, 114.53, 117.79, 119.29, 119.34, 121.57, 122.78, 127.46, 127.79, 129.29, 129.79, 133.31, 133.72, 136.98, 137.41, 139.12, 156.50, 161.50, 170.42.
Accurate mass analysis: m/z 639.3770, [MH]+, A = -0.2 ppm. Optical rotation: -27.36° @ 589 nm, concentration = 4.71 mg/mL in methanol. DESCRIPTION AND DOSAGE FORM
The physical and chemical properties of zavegepant (BHV-3500) drug substance mono-hydrochloride salt form are provided in Table 1.
Table 1 Physical and Chemical Properties
Biohaven number BHV-3500
Molecular formula C36H47CIN8O3
Molecular weight 675.26 (HO salt); 638.82 (free base)
Appearance White to off-white powder
Melting point ~178°C pH-solubility profile 105 mg/mL at pH = 8.2 and > 300 mg/mL at lower pH pKa 4.8 and 8.8 logD 1.21