














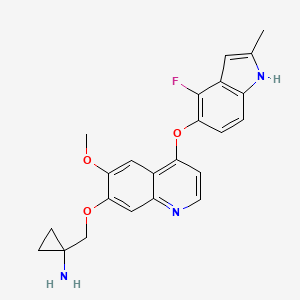
Catequentinib
C23H22FN3O3 407.4 g/mol
1-[[4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxyquinolin-7-yl]oxymethyl]cyclopropan-1-amine
1058156-90-3
CAS No. 1360460-82-7 DI HCL
| Molecular Weight | 480.36 |
| Formula | C23H22FN3O3 • 2HCl |
Anlotinib
AL3818
UNII-GKF8S4C432
Chia Tai Tianqing Pharmaceutical Group Co Ltd
Launched (Metastatic non small cell lung cancer – China – May-2018)
Orphan Drug; Priority Review
MOA:VEGFR inhibitor
Indication:advanced gastric adenocarcinoma; Advanced renal cell carcinoma (RCC); Medullary thyroid cancer (MTC); Metastatic colorectal cancer (CRC); Non small cell lung cancer (NSCLC); Soft tissue sarcoma; Ovarian cancerStatus:Phase III (Active)
AL-3818 ; AL-3818, Jiangsu Chia-tai Tianqing Pharmaceutical ; FOCUS-V ; FuKeWei ; VEGFR2/VEGFR3 inhibitor (capsule, cancer), Jiangsu Chia Tai Tianqing Pharmaceutical ; anlotinib ; anlotinib dihydrochloride ; catequentinib ; catequentinib ; catequentinib dihydrochloride
NMR https://file.selleckchem.com/downloads/nmr/S872601-Anlotinib-AL3818-hnmr-selleck.pdf
Anlotinib (AL3818) is a highly potent and selective VEGFR2 inhibitor with IC50 less than 1 nM. It has broad-spectrum antitumor potential in clinical trials.
Anlotinib dihydrochloride is in phase II/III clinical trials for the treatment of metastatic colorectal cancer and advanced gastric adenocarcinoma. The compound was co-developed by CTTQ Pharmaceutical (正大天晴) and Advenchen Laboratory.
It is also in phase II clinical trials for the treatment of ovarian cancer, endometrial cancer, non small cell lung cancer (NSCLC), medullary thyroid cancer (MTC), soft tissue sarcoma and advanced renal cell carcinoma (RCC).
In 2015, orphan drug designation was received in the U.S. for the treatment of ovarian cancer.
PATENT
WO 2016179123
https://patents.google.com/patent/WO2016179123A1/en
new process to synthesize l-((4-(4-Fluoro-2-methyl- lH- indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropanamine (AL3818) by condensing intermediate (XI) with (Yl) in a solvent at the presence of KI or Nal, or intermediate (X2) with (Y2) in a solvent to form intermediate (Z) which is deprotected to give the final compound (AL3818) in Scheme I. A stable crystalline form of l-((4-(4-Fluoro-2 -methyl- lH-indol-5-yloxy)-6- methoxyquinolin-7-yloxy)-methyl)cyclopropanamine and its salts as well as crystalline forms of salts have also been prepared.

Wherein, R is selected from H and Ci-Cealkoxy.
Process A

R is selected from H and C1 -C6 alkoxy
The final compound (AL3818) was prepared according to Process Al when R is H by deprotecting intermediate (Z-l) with HCOONH4 (ammonium formate) and Pd/C in an alcoholic solvent, such as MeOH, at 25°C-80°C for 0.1-4 hours. (Z-l) was prepared by reacting intermediate (XI) with (Yl-1) at the presence of KI or Nal with K2CO3 in a solvent, such as acetone or DMF, at a temperature of 60°C-160°C for 2-24 hours.
Process Al (R=H)

The final compound (AL3818) was prepared according to Process A2 when R is 4-OMe by deprotecting intermediate (Z-2) with TFA in DCM at 0°C-30°C for 1-24 hours. (Z-2) was prepared by reacting intermediate (XI) with (Y 1-2) at the presence of KI or Nal with K2C03 in a solvent, such as acetone or DMF, at a temperature of 60°C -160°C for 2-24 hours.
Process A2 (R=4-OMe)

The present invention relates a new process to synthesize l-((4-(4-Fluoro-2 -methyl- 1H- indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropanamine (AL3818) by reacting intermediate (X2) with (Y2) in a solvent to form intermediate (Z) which is deprotected to give the final compound (AL3818) according to Process B. Proce B

R is selected from H and C1-C6 alkoxy
The final compound (AL3818) was prepared according to Process Bl when R is H by deprotecting intermediate (Z-1) with HCOONH4 (ammonium formate) and Pd/C in an alcoholic solvent, such as MeOH, at 25°C-80°C for 0.1-4 hours. (Z-1) was prepared by reacting intermediate (X2-1) with (Y2) in a solvent, such as pyridine or lutidine, at a temperature of 60°C – 160°C for 1-12 hours.
Process Bl R=H)

The final compound (AL3818) was prepared according to Process B2 when R is 4-OMe by deprotecting intermediate (Z-2) with TFA in DCM at 0°C-30°C for 1-24 hours. (Z-2) was prepared by reacting intermediate (X2-2) with (Y2) in a solvent, such as pyridine or lutidine, at a temperature of 60°C -160°C for 1-12 hours.
Process B2 (R=4-OMe)

The following examples further illustrate the present invention, but should not be construed as in any way to limit its scope.
Example 1
Representation of Process A, Process Al
Process for preparation of l-((4-(4-Fluoro-2 -methyl- lH-indol-5-yloxy)-6-methoxy- quinolin-7-yloxy)methyl)cyclopropanamine (AL3818)
To a stirred mixture of benzyl l-(hydroxymethyl)cyclopropylcarbamate (50 g) and DCM (200 ml) was added DIPEA (39g). The result solution was cooled to 0-5 °C with ice/water and further stirred under this temperature for 15 min. MsCl (30g) was added via an addition funnel dropwise keeping temperature below 5°C for about 1.5 hours. After completion of addition, the reaction mixture was allowed stirring at 0-5°C for 30 min and quenched with saturated NaHC03 (150 ml). The solution was extracted with 150 ml DCM twice. The combined DCM layer was washed with 0.1 N HCl (400 ml) followed by brine. It was dried over Na2S04 and concentrated to obtain an off-white solid 60 gram as (l-(benzyloxycarbonylamino)cyclopropyl)methyl methanesulfonate (Yl-1), MS: (M+l) 300.
To a stirred mixture of (Yl-1) (16 g), XI [(4-(4-fluoro-2-methyl-lH-indol-5-yloxy)-6- methoxy-7-hydroxyquinoline, 12 g] , K2CO3 (21 g) and KI (21 g) was added DMF (100 ml), the reaction suspension was heated at 80°C for 10 hours and (Yl-l) (10 g) was added to continuously heated 80°C for 10 hours. The reaction then was quenched with water (150 ml) and extracted with 150 ml DCM twice. The combined DCM layer was washed with 2 N NaOH (100 ml) followed by water and brine. It was dried over Na2SC>4 and concentrated, further recrystallized from EtOH to obtain a yellow solid as benzyl l-((4-(4-fluoro-2-methyl-lH-indol-5-yloxy)-6-methoxyquinolin- 7-yloxy)methyl)cyclopropylcarbamate (Z-l) 9.5 g. MS: (M+l) 542.
To a stirred mixture of (Z-l) (9.5 g), HCOONH4 (4.7 g) and Pd/C (10%, wet 50%, 4.7g) was added MeOH, the reaction mixture was heated at 45°C for 1.5 hours. It was then cooled and filtered through Celite, further evaporated. 2N HCl (200 ml) was added and extracted with DCM/MeOH (10/1, 100 ml) twice. The aqueous layer was basified with 3N NaOH to adjust pH 11-12 to generate a solid precipitation. The solid was filtered and washed with water to neutral, further suction dry. The solid was dissolved into a mixture of DCM/MeOH (250 ml, 10/1) and further washed with water and brine. It was dried with MgS04 and filtered, further evaporated to give a light yellow solid 5.5 g crude product. Further purification was conducted by dissolving the crude product into DCM/MeOH (40 ml, 10/1) to triturate with petroleum ether (40 ml) for 2 hours slow stirring. The precipitate was filtered and dried in an oven to give the final crystalline product 4.4 g (MP: 203-208 C) and it can be further purified by recrystallizing from EtOH to give purer final product as a same crystalline form. MS: (M+l) 408; ¾ NMR(DMSO-dg) δ 0.60- 0.63(d, 4H), 2.41(s, 1H), 2.42-2.5 l(t, 2H), 3.3 l(s, 2H), 3.96(s, 3H), 4.04(s, 2H), 6.27(s, 1H), 6.31-6.32(m, 1H), 6.97-7.02(t, 1H), 7.20-7.22(d, 1H), 7.36(s, 1H), 7.60(s, 1H), 8.40-8.42(d, 1H), 1 1.41(s, 1H). MP: 208-210°C; DSC Melting Range (Endo): 207-220°C with Peak Temp=216°C

-
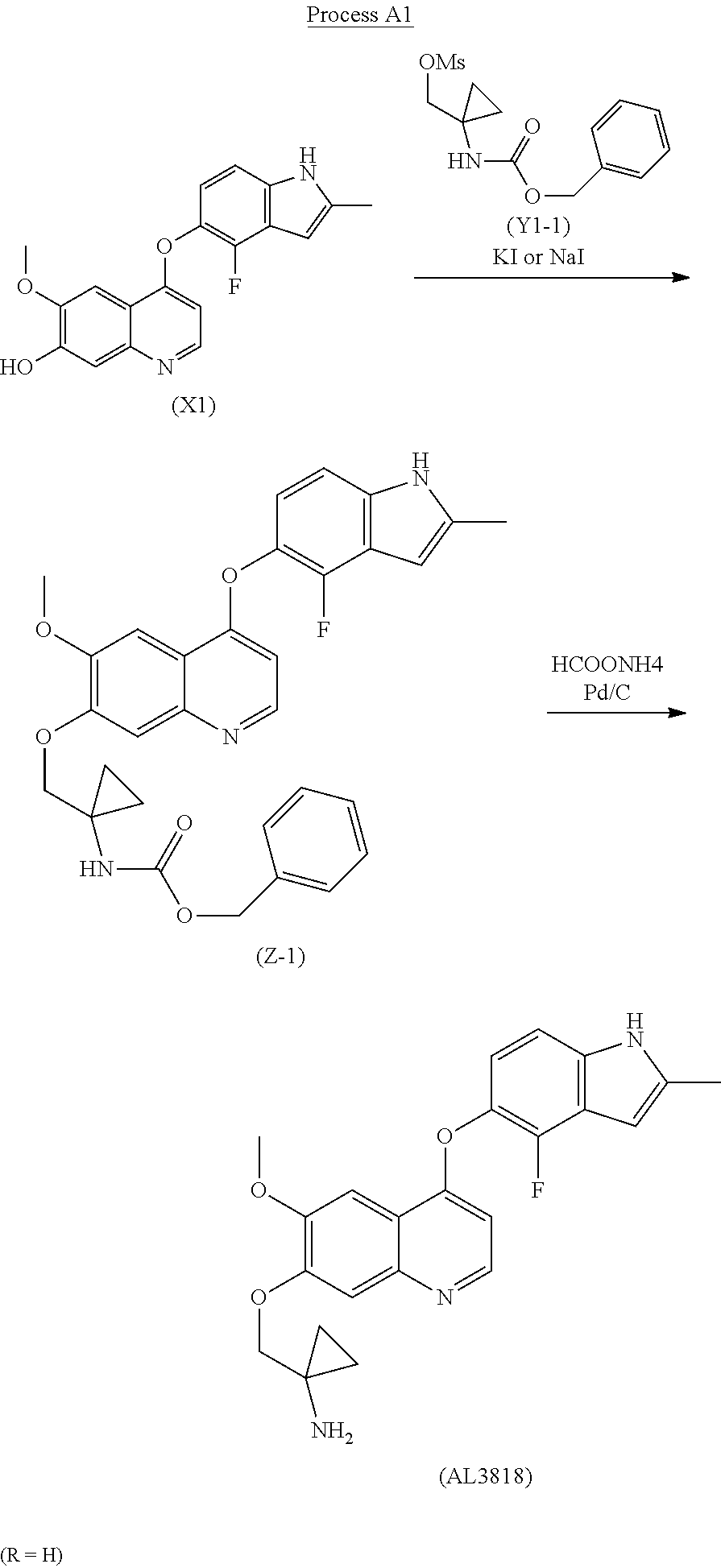
The present invention relates a new process to synthesize 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropanamine (AL3818) by condensing intermediate (X1) with (Y1) in a solvent at the presence of KI or NaI to form intermediate (Z) which is deprotected to give the final compound (AL3818) according to Process A.
-

- [0040]
The final compound (AL3818) was prepared according to Process A1 when R is H by deprotecting intermediate (Z-1) with HCOONH4 (ammonium formate) and Pd/C in an alcoholic solvent, such as MeOH, at 25° C.-80° C. for 0.1-4 hours. (Z-1) was prepared by reacting intermediate (X1) with (Y1-1) at the presence of KI or NaI with K2CO3 in a solvent, such as acetone or DMF, at a temperature of 60° C.-160° C. for 2-24 hours.
-
- [0041]
The final compound (AL3818) was prepared according to Process A2 when R is 4-OMe by deprotecting intermediate (Z-2) with TFA in DCM at 0° C.-30° C. for 1-24 hours. (Z-2) was prepared by reacting intermediate (X1) with (Y1-2) at the presence of KI or NaI with K2CO3 in a solvent, such as acetone or DMF, at a temperature of 60° C.-160° C. for 2-24 hours.
-

- [0042]
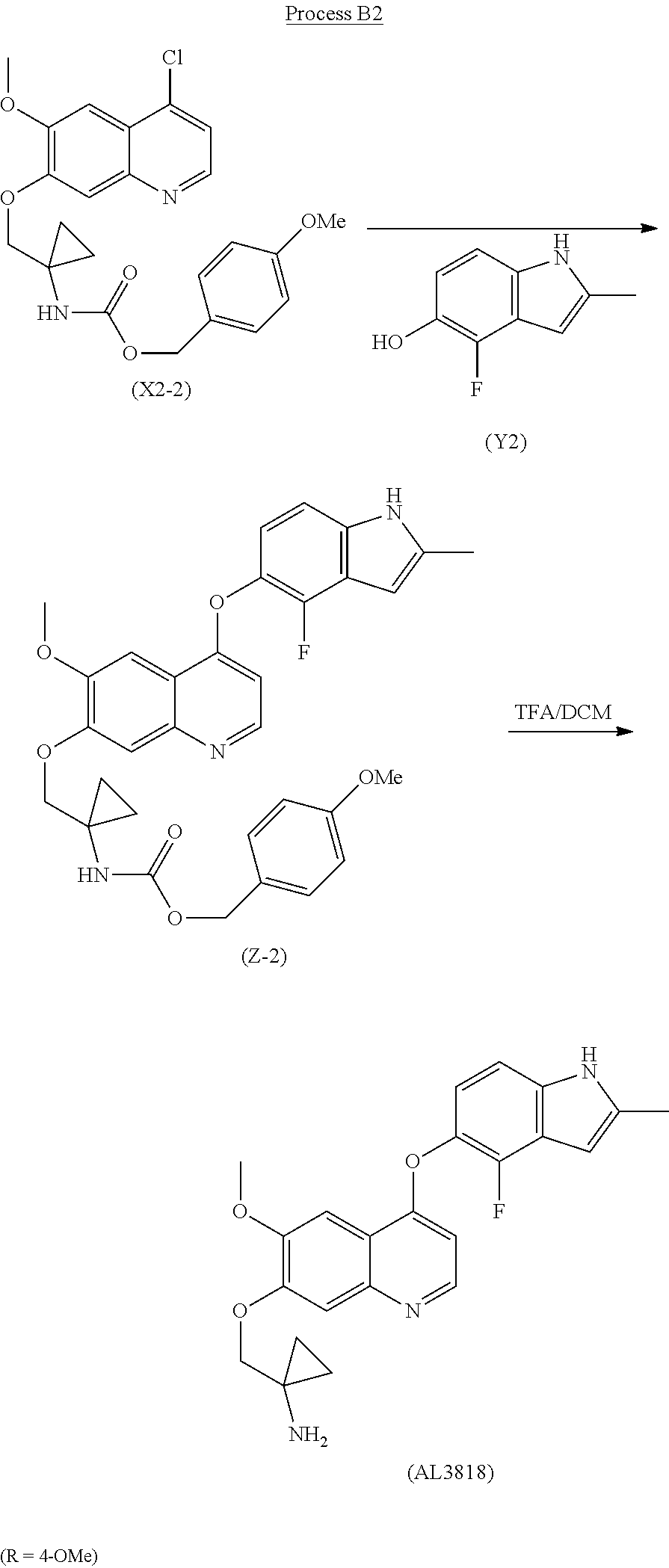
The present invention relates a new process to synthesize 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropanamine (AL3818) by reacting intermediate (X2) with (Y2) in a solvent to form intermediate (Z) which is deprotected to give the final compound (AL3818) according to Process B.
-

- [0043]
The final compound (AL3818) was prepared according to Process B1 when R is H by deprotecting intermediate (Z-1) with HCOONH4 (ammonium formate) and Pd/C in an alcoholic solvent, such as MeOH, at 25° C.-80° C. for 0.1-4 hours. (Z-1) was prepared by reacting intermediate (X2-1) with (Y2) in a solvent, such as pyridine or lutidine, at a temperature of 60° C.-160° C. for 1-12 hours.
-

- [0044]
The final compound (AL3818) was prepared according to Process B2 when R is 4-OMe by deprotecting intermediate (Z-2) with TFA in DCM at 0° C.-30° C. for 1-24 hours. (Z-2) was prepared by reacting intermediate (X2-2) with (Y2) in a solvent, such as pyridine or lutidine, at a temperature of 60° C.-160° C. for 1-12 hours.
-
- [0045]
The following examples further illustrate the present invention, but should not be construed as in any way to limit its scope.
Example 1Representation of Process A, Process A1Process for preparation of 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-quinolin-7-yloxy)methyl)cyclopropanamine (AL3818)
- [0046]
To a stirred mixture of benzyl 1-(hydroxymethyl)cyclopropylcarbamate (50 g) and DCM (200 ml) was added DIPEA (39 g). The result solution was cooled to 0-5° C. with ice/water and further stirred under this temperature for 15 min. MsCl (30 g) was added via an addition funnel dropwise keeping temperature below 5° C. for about 1.5 hours. After completion of addition, the reaction mixture was allowed stirring at 0-5° C. for 30 min and quenched with saturated NaHCO3 (150 ml). The solution was extracted with 150 ml DCM twice. The combined DCM layer was washed with 0.1 N HCl (400 ml) followed by brine. It was dried over Na2SO4 and concentrated to obtain an off-white solid 60 gram as (1-(benzyloxycarbonylamino)cyclopropyl)methyl methanesulfonate (Y1-1), MS: (M+1) 300.
- [0047]
To a stirred mixture of (Y1-1) (16 g), X1 [(4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-7-hydroxyquinoline, 12 g], K2CO3 (21 g) and KI (21 g) was added DMF (100 ml), the reaction suspension was heated at 80° C. for 10 hours and (Y1-1) (10 g) was added to continuously heated 80° C. for 10 hours. The reaction then was quenched with water (150 ml) and extracted with 150 ml DCM twice. The combined DCM layer was washed with 2 N NaOH (100 ml) followed by water and brine. It was dried over Na2SO4 and concentrated, further recrystallized from EtOH to obtain a yellow solid as benzyl 1-((4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropylcarbamate (Z-1) 9.5 g. MS: (M+1) 542.
- [0048]
To a stirred mixture of (Z-1) (9.5 g), HCOONH4 (4.7 g) and Pd/C (10%, wet 50%, 4.7 g) was added MeOH, the reaction mixture was heated at 45° C. for 1.5 hours. It was then cooled and filtered through Celite, further evaporated. 2N HCl (200 ml) was added and extracted with DCM/MeOH (10/1, 100 ml) twice. The aqueous layer was basified with 3N NaOH to adjust pH 11-12 to generate a solid precipitation. The solid was filtered and washed with water to neutral, further suction dry. The solid was dissolved into a mixture of DCM/MeOH (250 ml, 10/1) and further washed with water and brine. It was dried with MgSO4 and filtered, further evaporated to give a light yellow solid 5.5 g crude product. Further purification was conducted by dissolving the crude product into DCM/MeOH (40 ml, 10/1) to triturate with petroleum ether (40 ml) for 2 hours slow stirring. The precipitate was filtered and dried in an oven to give the final crystalline product 4.4 g (MP: 203-208° C.) and it can be further purified by recrystallizing from EtOH to give purer final product as a same crystalline form. MS: (M+1) 408; 1H NMR (DMSO-d6) δ 0.60-0.63 (d, 4H), 2.41 (s, 1H), 2.42-2.51 (t, 2H), 3.31 (s, 2H), 3.96 (s, 3H), 4.04 (s, 2H), 6.27 (s, 1H), 6.31-6.32 (m, 1H), 6.97-7.02 (t, 1H), 7.20-7.22 (d, 1H), 7.36 (s, 1H), 7.60 (s, 1H), 8.40-8.42 (d, 1H), 11.41 (s, 1H). MP: 208-210° C.; DSC Melting Range (Endo): 207-220° C. with Peak Temp=216° C. TGA demonstrating as an unsolvated material with weight loss at about 210° C. (between 205-215° C.). XRPD having pattern comprising characteristic 10 peaks with intensity % greater than 10% expressed in d values and angles as follows:
-
Angle d value 13.344 6.62986 15.858 5.58405 16.799 5.27326 17.640 5.02377 18.770 4.72373 20.650 4.29771 21.633 4.10463 23.087 3.84934 25.128 3.54112 26.607 3.34755
- [0049]
It was similar prepared according to the preparation procedures of (Z-1) described in Example 1 by using 4-methoxybenzyl 1-(hydroxymethyl)cyclopropylcarbamate to first generate (1-((4-methoxybenzyloxy)carbonylamino)cyclopropyl)methyl methanesulfonate (Y1-2) then to give 4-methoxybenzyl 1-((4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)-methyl)cyclopropylcarbamate (Z-2), MS: (M+1) 572
- [0050]
To a stirred mixture of (Z-2) (1.5 g) in DCM (15 ml) at 0° C. was added TFA (1.5 ml) for about 30 min and warmed up to RT. The reaction was stirred at RT for 2 hours and added into water (30 ml). The aqueous layer was extracted with DCM twice (100 ml×2) and basified with 2N NaOH to adjust pH 11-12. The mixture was extracted with DCM (100 ml×3) and further washed with brine (100 ml). It was dried with MgSO4 and filtered. The solution was evaporated to give 1.05 g crude final product. Further purification was conducted to dissolve the crude product into DCM/MeOH and triturated with petroleum ether and dried in an oven to give the final pure product 0.8 g AL3818 with the same crystalline form.
- Example 2Representation of Process A, Process A2Process for preparation of 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-quinolin-7-yloxy)methyl)cyclopropanamine (AL3818)
Example 3Representation of Process A, Process B1Process for preparation of 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-quinolin-7-yloxy)methyl)cyclopropanamine (AL3818)
- [0051]
To a mixture of benzyl 1-((4-chloro-6-methoxyquinolin-7-yloxy)methyl)cyclopropyl-carbamate (X2-1) (5 g), 4-fluoro-2-methyl-1H-indol-5-ol (Y2) (5 g) and DMAP (4 g) was added 1,6-lutidine (15 ml). The reaction was stirred and heated at 135° C. for 5 hours and was cooled followed by adding IPA with slow stirring for 2 hours at RT. The solid was filtered and further washed with IPA, dried to give (Z-1) 5.2 g as a solid. It was then similarly prepared according to deprotection procedures described of (Z-1) in Example 1 to give the final compound AL3818 with the same crystalline form.
Example 4Representation of Process A, Process B2Process for preparation of 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-quinolin-7-yloxy)methyl)cyclopropanamine (AL3818)
- [0052]
(Z-2) was similarly prepared according to the procedures described in Example 3 by using 4-methoxybenzyl 1-((4-chloro-6-methoxyquinolin-7-yloxy)methyl)cyclopropylcarbamate (X2-2) and (Y2). It was then similarly prepared according to deprotection procedures of (Z-2) described in Example 2 to give the final compound AL3818 with the same crystalline form.
Example 5
- [0053]
Preparation of 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-quinolin-7-yloxy)-methyl)cyclopropanamine bishydrochloride acid salt and its crystalline
- [0054]
To a 25 ml flask was added 250 mg free base (AL3818), 4N HCl in dioxane 0.625 mL (2.5 mmol, 4 eq.) in 10 ml EtOH, the reaction was heated at 75° C. for 30 minutes, cooled to RT and stirred for O.N. The solid was filtered and rinsed with acetone twice. It was dried in oven at 50° C. for 4 hours to give 126 mg white solid as the bishydrochloride salt as a crystalline and further recrystallized from EtOH to give a purer product as a same crystalline form. 1H NMR (DMSO-d6) δ 1.09-1.24 (m, 4H), 2.43 (s, 3H), 4.08 (s, 3H), 4.40 (s, 2H), 6.32 (s, 1H), 6.76 (s, 1H), 7.05-7.11 (t, 1H), 7.27-7.30 (d, 1H), 7.65 (s, 1H), 7.82 (s, 1H), 8.64 (s, 2H), 8.70-8.73 (m, 1H), 11.51 (s, 1H). Chloride ion chromatography showed 2 molecular ratio ions (16.1%). DSC Melting Range (Exo): 249-280 with Peak Temp=268° C.
- Example 6Preparation of 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-quinolin-7-yloxy)methyl)cyclopropanamine bishydrochloridehydrate acid salt and its crystalline
- [0055]
To a 10 mL flask, charged 140 mg of 3818-2HCl salt from above Example 4 and 0.7 mL (×5 with salt volume) of 80% MeOH in H2O. The result suspension was heated to 70° C. to form a solution and cooled to RT and further stirred for O.N. The solid was filtered and rinsed with acetone twice. It was dried in oven at 50° C. for 4 hours to obtain off-white solid 110 mg as the crystalline bishydrochloride hydrate salt. 1H NMR (DMSO-d6) δ 1.09 (s, 2H), 1.22 (s, 2H), 2.44 (s, 1H), 2.52 (s, 2H), 4.09 (s, 3H), 4.44 (s, 2H), 6.32 (s, 1H), 6.81-6.82 (d, 1H), 7.08-7.14 (t, 1H), 7.29-7.32 (d, 1H), 7.79 (s, 1H), 7.85 (s, 1H), 8.75-8.78 (d, 1H), 8.85 (s, 2H), 11.66 (s. 1H). Chloride ion chromatography showed 2 molecular ratio ions (17.8%). DSC Melting Range (Exo): 207-260° C. with Peak Temp=226° C. TGA demonstrating 2.68% (˜3%, 1 water) weight loss till 120° C. (between 115-125° C.) and further weight loss at about 170° C. (between 165-175° C.).

/////////////catequentinib, ANLOTINIB, AL3818, AL 3818, PHASE 2, CHINA 2018
NC1(CC1)COc1cc2nccc(Oc3ccc4[NH]c(C)cc4c3F)c2cc1OC