














 |
|
| IUPAC Condensed | cyclo[Arg-Gly-Asp-D-Phe-N(Me)Val] |
|---|---|
| HELM | PEPTIDE1{R.G.D.[dF].[meV]}$PEPTIDE1,PEPTIDE1,5:R2-1:R1$$$ |
| IUPAC | cyclo[L-arginyl-glycyl-L-alpha-aspartyl-D-phenylalanyl-N-methyl-L-valyl] |
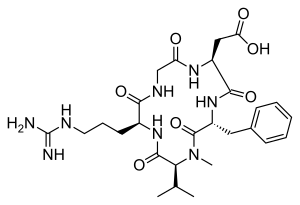
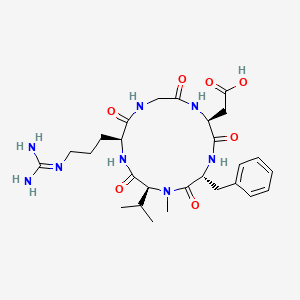
CILENGITIDE
- Molecular FormulaC27H40N8O7
- Average mass588.656 Da
2-[(2S,5R,8S,11S)-5-benzyl-11-[3-(diaminomethylideneamino)propyl]-7-methyl-3,6,9,12,15-pentaoxo-8-propan-2-yl-1,4,7,10,13-pentazacyclopentadec-2-yl]acetic acid
Cilengitide has been in phase III clinical trials by Merck Serono and NCI for the treatment of glioblastoma multiforme. However, this research has been discontinued.
Cilengitide was originally developed by Merck KGaA in collaboration with the Technical University of Munich, then received orphan drug designation from FDA for the treatment of glioma in 2005.
Cilengitide (EMD 121974) is a molecule designed and synthesized at the Technical University Munich in collaboration with Merck KGaA in Darmstadt. It is based on the cyclic peptide cyclo(-RGDfV-), which is selective for αv integrins, which are important in angiogenesis (forming new blood vessels), and other aspects of tumor biology. Hence, it is under investigation for the treatment of glioblastoma, where it may act by inhibiting angiogenesis, and influencing tumor invasion and proliferation.[1][2]
The European Medicines Agency has granted cilengitide orphan drug status.[3]
Cilengitide seems to function by inhibiting the FAK/src/AKT pathway and inducing apoptosis in endothelial cells.[4] Preclinical studies in mice of cilengitide were able to demonstrate efficacious tumor regression.[4]
In a rat xenograft model, cilengitide was able to potentiate the cytotoxic effects of radiation when cilengitide was administered prior to radiation therapy.[5] When combined with radiation, inhibition of integrin expression by cilengitide synergistically improves the cytotoxic effects of ionizing radiation for glioblastoma.[5]
Clinical trials
Phase II studies were able to demonstrate that cilengitide as a potential monotherapy in patients with recurrent glioblastoma[6] with high intratumor drug levels when 2000 mg of cilengitide is given twice weekly.[7]
Cilengitide is well tolerated, in combination with radiation and temozolomide, at a dose of 2000 mg in patients with newly diagnosed glioblastoma, regardless of MGMT promoter status.[8] In a phase I/IIa study, the addition of cilengitide to the standard of care for newly diagnosed glioblastoma (surgical resection followed by temozolomide and radiation therapy) improves progression-free survival and overall survival in patients with MGMT promoter methylation.[9]
However, in a subsequent study, cilengitide does not seem to alter the pattern of glioblastoma progression,[10]
and in an EORTC phase III randomized, controlled, multicenter clinical trial, consisting of over 500 patients in 23 countries, the addition of cilengitide to the standard of care did not improve overall survival in patients with newly diagnosed glioblastoma and methylated MGMT promoter status [11] A phase II study, the CORE trial, is currently being conducted in patients with newly diagnosed glioblastoma and unmethylated MGMT promoter status.[12]

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN
Angewandte Chemie, International Edition, 55(4), 1540-1543; 2016
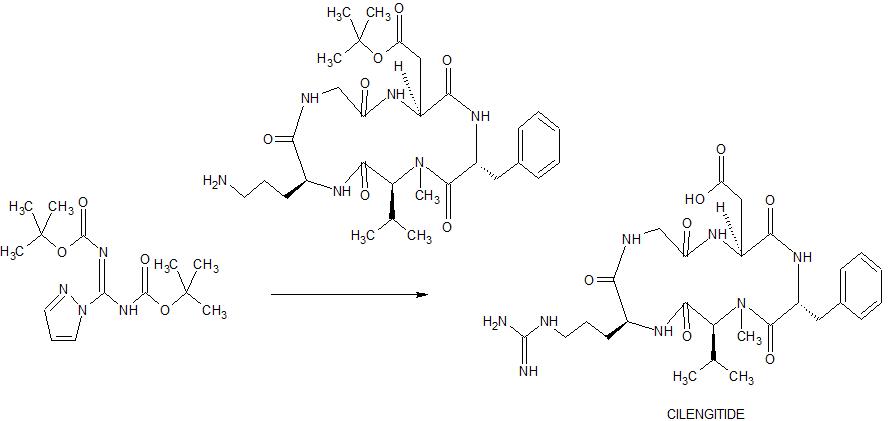
SYN
Chemistry – A European Journal, 16(18), 5385-5390, S5385/1-S5385/36; 2010
1. WO0047228A1 / US7115261B1.
2. US6001961A.

1. CN102731627A.

Peptide purification. The end product 3 was dissolved in 5 ml MilliQ water and it was filtered through a 0.2 pm filter. The cyclic peptide was purified by semipreparative RP-HPLC using acetronitrile (0.05% TFA)/water (0.1 % TFA). The HPLC sample was vacuum concentred and transformed into the hydrochloride salt lyophilized with water with 0.05% HCI.1H-NMR (500 MHz, H20:D20-d2 9: 1 , 278 K): δ = 9.29 (t, NH Gly), 9.20 (d, J = 7.24 Hz, NH Asp), 8.90 (t, J = 5.89/5.89 Hz, NH Thz), 8.46 (d, J = 8.93 Hz, NH Arg), 7.79 (s, CH Thz), 7.22 (t, J = 5.39/5.39 Hz, ΝΗε Arg), 4.75 (m, CHa Arg), 4.63 (m, CHa Asp), 4.04 (dd, J = 3.35/14.90 Hz, CHa Gly), 3.82 (dd, J = 6.69/14.96 Hz, CHa Gly), 3.17 (m, CH25 Arg), 2.89 (m, CH2p Asp), 1 .92 (m, CH p Arg), 1 .82 (m, CHP Arg), 1 .63 (m, CH2 Arg). HPLC (gradient 0 to 20% of CH3CN in 15 min): tR= 10.52 m in. HRMS (E IS) m/z calculated 468.1540
found 469.16099 (M+H)+.
References
- ^ Burke PA, DeNardo SJ, Miers LA, Lamborn KR, Matzku S, DeNardo GL (August 2002). “Cilengitide targeting of alpha(v)beta(3) integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts”. Cancer Research. 62 (15): 4263–72. PMID 12154028.
- ^ Goodman SL, Hölzemann G, Sulyok GA, Kessler H (February 2002). “Nanomolar small molecule inhibitors for alphav(beta)6, alphav(beta)5, and alphav(beta)3 integrins”. Journal of Medicinal Chemistry. 45 (5): 1045–51. doi:10.1021/jm0102598. PMID 11855984.
- ^ Spreitzer H (October 27, 2008). “Neue Wirkstoffe – Cilengitide”. Österreichische Apothekerzeitung (in German) (22/2008): 1136–7.
- ^ Jump up to:a b Yamada S, Bu XY, Khankaldyyan V, Gonzales-Gomez I, McComb JG, Laug WE (December 2006). “Effect of the angiogenesis inhibitor Cilengitide (EMD 121974) on glioblastoma growth in nude mice”. Neurosurgery. 59 (6): 1304–12, discussion 1312. doi:10.1227/01.NEU.0000245622.70344.BE. PMID 17277694. S2CID 19861713.
- ^ Jump up to:a b Mikkelsen T, Brodie C, Finniss S, Berens ME, Rennert JL, Nelson K, Lemke N, Brown SL, Hahn D, Neuteboom B, Goodman SL (June 2009). “Radiation sensitization of glioblastoma by cilengitide has unanticipated schedule-dependency”. International Journal of Cancer. 124 (11): 2719–27. doi:10.1002/ijc.24240. PMID 19199360.
- ^ Reardon DA, Fink KL, Mikkelsen T, Cloughesy TF, O’Neill A, Plotkin S, et al. (December 2008). “Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme”. Journal of Clinical Oncology. 26 (34): 5610–7. CiteSeerX 10.1.1.688.8987. doi:10.1200/JCO.2008.16.7510. PMID 18981465.
- ^ Gilbert MR, Kuhn J, Lamborn KR, Lieberman F, Wen PY, Mehta M, Cloughesy T, Lassman AB, Deangelis LM, Chang S, Prados M (January 2012). “Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03-02, a phase II trial with measures of treatment delivery”. Journal of Neuro-Oncology. 106 (1): 147–53. doi:10.1007/s11060-011-0650-1. PMC 4351869. PMID 21739168.
- ^ Nabors LB, Mikkelsen T, Hegi ME, Ye X, Batchelor T, Lesser G, Peereboom D, Rosenfeld MR, Olsen J, Brem S, Fisher JD, Grossman SA (November 2012). “A safety run-in and randomized phase 2 study of cilengitide combined with chemoradiation for newly diagnosed glioblastoma (NABTT 0306)”. Cancer. 118 (22): 5601–7. doi:10.1002/cncr.27585. PMC 3423527. PMID 22517399.
- ^ Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, Clement PM, et al. (June 2010). “Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma” (PDF). Journal of Clinical Oncology. 28(16): 2712–8. doi:10.1200/JCO.2009.26.6650. PMID 20439646.
- ^ Eisele G, Wick A, Eisele AC, Clément PM, Tonn J, Tabatabai G, et al. (March 2014). “Cilengitide treatment of newly diagnosed glioblastoma patients does not alter patterns of progression”(PDF). Journal of Neuro-Oncology. 117 (1): 141–5. doi:10.1007/s11060-014-1365-x. PMID 24442484. S2CID 21636884.
- ^ Merck Group. “Phase III Trial of Cilengitide Did Not Meet Primary Endpoint in Patients With Newly Diagnosed Glioblastoma, Date accessed: 3/24/2014.”
- ^ ASCO Meeting Library. [1] “Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma and methylated O6-methylguanine-DNA methyltransferase (MGMT) gene promoter: Key results of the multicenter, randomized, open-label, controlled, phase III CENTRIC study, Date accessed: 3/24/2014
 |
|
| Names | |
|---|---|
| IUPAC name
2-[(2S,5R,8S,11S)-5-benzyl-11-{3-[(diaminomethylidene)amino]propyl}-7-methyl-3,6,9,12,15-pentaoxo-8-(propan-2-yl)-1,4,7,10,13-pentaazacyclopentadecan-2-yl]acetic acid
|
|
| Identifiers | |
|
|
|
3D model (JSmol)
|
|
| ChEMBL | |
| ChemSpider | |
| KEGG | |
| MeSH | Cilengitide |
|
PubChem CID
|
|
| UNII | |
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C27H40N8O7 | |
| Molar mass | 588.656 g/mol |
| Density | 1.417 g/mL |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Infobox references | |
/////////CILENGITIDE, циленгитид , سيلانجيتيد ,西仑吉肽 , PHASE 3, EMD 121974, EMD-121974, UNII-4EDF46E4GI, orphan drug , MERCK, glioblastoma,
CC(C)C1C(=O)NC(C(=O)NCC(=O)NC(C(=O)NC(C(=O)N1C)CC2=CC=CC=C2)CC(=O)O)CCCN=C(N)N