














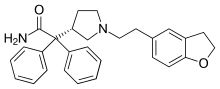
Darifenacin
2-[(3S)-1-[2-(2,3-dihydro-1-benzofuran-5-yl)ethyl]pyrrolidin-3-yl]-2,2-diphenylacetamide
Darifenacin; Emselex; Enablex; CAS 133099-04-4; UNII-APG9819VLM;
US 2004-12-22 APPROVED
EU 2004-10-22 APPROVED
| Molecular Formula: | C28H30N2O2 |
|---|---|
| Molecular Weight: | 426.56 g/mol |
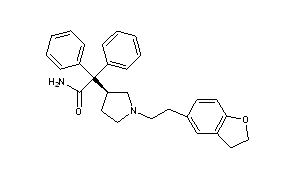
Derivative Type: Hydrobromidd
CAS Registry Number: 133099-07-5
Research Code:UK-88525-04
Trade Name:Emselex® / Enablex® / Xelena®
MOA:M3 muscarinic acetylcholine receptor antagonistIndication:Overactive bladder (OAB)
Status:Approved
Company:Novartis (Originator) , Merus Labs,Warner chilcottSales:
ATC Code:G04BD10
臭化水素酸ダリフェナシン
Darifenacin Hydrobromide

C28H30N2O2▪HBr : 507.46
[133099-07-7]
Darifenacin (originally developed by Pfizer, trade name En`blex in USA and Canada, Emselex in Europe) is an effective medibatinn used for treatment of overactive bladder (OAB) symptoms.
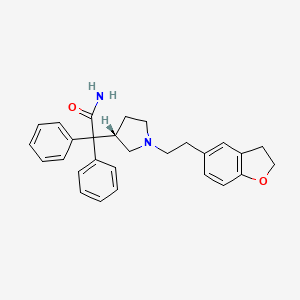
OAB is a common condition symptomized by urinary urgency, with or without urge in continence, usually with frequency and nocturia that notably affects the lives of millions of people. Human bladder tissue contains M2 (80%) and M3 (20%) muscarinic receptors, and the latter act as the primary mediator of detrusor contraction in response to cholinergic activation.
So muscarinic receptor antagonists are the current treatment of choice for OAB. As different subtypes of muscarinic receptors are widely distributed in the human body to play key physiological roles, a very selective M3 receptor antagonist is in high demand in the market for OAB medication. Darifenacin is a potent and competitive M3 selective receptor antagonist (M3SRA) that has been shown to have high affinity and selectivity (59-fold higher) for the M3 receptor, with low selectivity for the other muscarinic receptor subtypes. Its hydrobromide salt is the active ingredient of pharmaceutical formulations. The efficacy, tolerability and safety of darifenacin in the treatment of OAB are well established.
Darifenacin (trade name Enablex in US and Canada, Emselex in Europe) is a medication used to treat urinary incontinence. It was discovered by scientists at the Pfizer research site in Sandwich, UK under the identifier UK-88,525 and used to be marketed by Novartis. In 2010 the US rights were sold to Warner Chilcott for 400 million US$.
Mechanism of action
Darifenacin works by blocking the M3 muscarinic acetylcholine receptor, which is primarily responsible for bladder muscle contractions. It thereby decreases the urgency to urinate. It is not known whether this selectivity for the M3 receptor translates into any clinical advantage when treating symptoms of overactive bladder syndrome.
It should not be used in people with urinary retention. Anticholinergic agents, such as darifenacin, may also produce constipation and blurred vision. Heat prostration (due to decreased sweating) can occur when anticholinergics such as darifenacin are used in a hot environment.[1]
Clinical uses
Darifenacin is indicated for the treatment of overactive bladder with symptoms of urge urinary incontinence, urgency and frequency in adults.
clip
http://nopr.niscair.res.in/bitstream/123456789/18844/1/IJCb%2052B(6)%20824-828.pdf
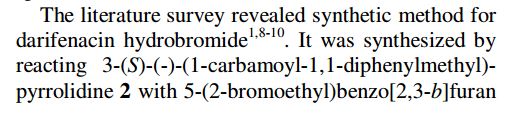

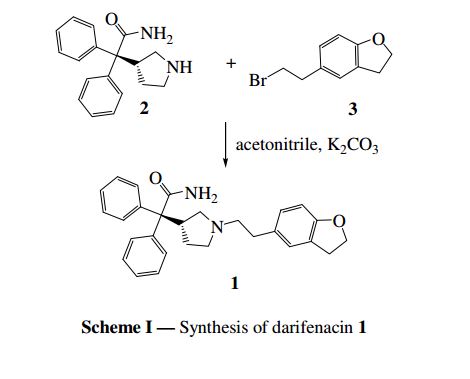
The substance was first described in EP 388 054. The method of its preparation in accordance with this document is shown in the following scheme.
Scheme 1

DARIFENACIN

wherein the substituents R and X can be

A particular preferable embodiment is shown in Scheme 2, wherein substance VII is alkylated with 5-(2-bromoethyl)-2,3-dihydrobenzofuran (VIII) in the presence of potash by reflux in acetonitrile. Crude darifenacin (IX) is purified using column chromatography and crystallized from diisopropylether
Scheme 2

Scheme 2: Synthesis of darifenacin by N-alkylation of pyrrolidine VII with 5-(2-bromoethyi)-
2,3-dihydrobenzofuran (VIII)
Darifenacin hydrobromide is prepared by precipitation of purified darifenacin base dissolved in acetone by addition of concentrated aqueous HBr.
However, in repeated reproduction these procedures did not provide a product of an adequate quality in a reasonable industrially applicable yield. It has been found out that a portion of the resulting darifenacin undergoes subsequent alkylation to the second stage, producing the twice substituted substance X. In the course of the reaction undesired reactions of 5-(2-biOmoethyl)-2,3-dihydrobenzofuran VIII also occur, namely hydrolysis producing a hydroxy derivative (XI) and elimination producing a vinyl derivative (XII). All these reactions reduce the yield of the desired substance and complicate the preparation of high-quality API.
By reproduction of the above mentioned procedure a substance was obtained with the following contents of constituents in accordance with HPLC [%] : VII 2.8 VIII 14.2 1X 57.2 X 7.8 XI 1.2 XII 8.0.

A purification procedure for darifenacin was published in WO03080599A1.
Darifenacin in t-amyl alcohol is heated with Amberlite (22 h), the solid fraction is filtered off, the solvent is evaporated from the filtrate and the residue is dissolved in toluene; a solvate of darifenacin with toluene is separated by cooling. This solvate can be directly used for the preparation of darifenacin hydrobromide (the solvate is dissolved in 2-butanol, concentrated HBr is added and the darifenacin salt is separated by cooling).
Another method of purification of darifenacin, described in the same document, is conversion of the darifenacin/toluene solvate to darifenacin hydrate (the solvate is dissolved in acetonitrile and water is added under gradual separation of darifenacin hydrate (Scheme 3)), which can be used for the preparation of salts or can be directly incorporated into pharmaceutical forms. The hydrate can be optionally converted to the hydrogen bromide in a similar way as the solvate.

(IX.W)
Scheme 3: Methods of purification of crude darifenacin and its conversion to hydrobromide
During reproduction of the purification procedure it was possible to separate a portion of substance X in the solid phase form after dissolution of crude darifenacin in toluene. However, the attempt to obtain the desired toluene solvate of darifenacin from the toluene solution was not successful during the reproduction. This means that this method does not lead to the pure substance.
WO2007076159 (TEVA) describes preparation of darifenacin from dihydrobenzofuran ethylchloride and carbamoyl(diphenylmethyl)pyrrolidine tartrate in the aqueous phase using K2CO3 as the base. After cooling of the reaction mixture n-butanol is added, the aqueous and organic phases are separated, acetanhydride is added and a reaction with concentrated hydrobromic acid (48%) is performed.
This method enables preparation of the substance with a satisfactory yield, ca. 77%; however, the reaction in the aqueous phase takes place in the melt, which is very thick, which causes techno logical problems, e.g. difficult stirring, sticking of the mixture on the walls of the reaction vessel, etc. During a reproduction of this procedure it was found that acetanhydride caused partial decomposition of the product and formation of further impurities. The crude product prepared this way cannot be converted to hydrobromide without further purification. N-butanol mentioned in the procedure is partly miscible with water, which also has a negative impact on the process yield. Contents of constituents (HPLC [%]) in the crude product within the reproduction of the procedure in accordance with WO2007076159 (TEVA):
Reaction with dihydrobenzofuran ethylchloride: VII 1.9 VIII 6.1 1X 82.0 X 6.3 XI not found XII not found
Reaction with dihydrobenzofuran ethylbromide: VII 2.8 VIII 0.5 1X 77.5 X 9.5 XI 2.0 XII 2.4
The above mentioned analysis of the described procedures and attempts to reproduce them have revealed that compound X is the major problem. During the application it was never possible to obtain the product that would contain less than 5% of this impurity. The substance is similar to the desired product in its character, it has similar solubility in most solvents and moreover it also changes to hydrogen bromide or other salts. For this reason it is very difficult to separate this substance by normal crystallization of the base or one of the salts of darifenacin.
While toluene has proved suitable for this function in the above-described procedures (WO03080599A1), after the separation of a portion of substance X it was not possible to obtain the desired toluene solvate of darifenacin. The procedure appears to be hardly usable without further modification and it does not lead to the desired pure product.
Darifenacin (la) is chemically known as (S)-2-[l-[2-(2,3-Dihydrobenzofuran-5- yl)ethyl]-3-pyrrolidinyl]-2,2-diphenylacetamide and is approved as hydrobromide salt. Darifenacin is a potent muscarinic M3 receptor antagonist. Muscarinic receptors play an important role in several major cholinergically mediated functions, including contractions of the urinary bladder, gastrointestinal smooth muscle, saliva production, and iris sphincter function. Darifenacin has greater affinity for the M receptor than for the other known muscarinic receptors. Darifenacin hydrobromide is commercially available under the brand name Enablex® in the US. It has been approved for the treatment of overactive bladder with symptoms of urge urinary incontinence, urgency and frequency.
US 5,096,890 disclosed Darifenacin and its pharmaceutically acceptable salts. US ‘890 discloses several processes for preparing Darifenacin. According to the process disclosed in US ‘890, Darifenacin (la) may be prepared by condensing 5-(2-bromoethyl)-2,3-dihydrobenzofuran (II) with 3-(S)-(-)-(l – carbamoyl-l , l -diphenylmethyl)pyrrolidine (III) in the presence of K2C03 in acetonitrile.
The process is as shown in Scheme-I below:
1. Anhydrous K2C03

US ‘890 also discloses a variant process for the preparation of Darifenacin (la) by condensing 5-(2-bromoethyl)-2,3-benzofuran (IV) with 3-(S)-(-)-(l -carbamoyl- 1 , 1- diphenylmethyl)pyrrolidine (III) in the presence of K2C03 in acetonitrile to produce (S)-2-[l-[2-(2,3-benzofuran-5-yl)ethyl]-3-pyrrolidinyl]-2,2-diphenylacetamide (V), which is further hydrogenated in the presence of Pd/C in acetic acid to produce Darifenacin crude, followed by purification using column chromatography.
The rocess is as shown in Scheme-II below:

Darifenacin
(la)
US ‘890 also discloses an another variant process for the preparation of Darifenacin hydrobromide (I) by condensing 5-chloroacetyl-2,3-dihydrobenzofuran (VI) with 3- (S)-(-)-(l-carbamoyl-l ,l-diphenylmethyl)pyrrolidine (III) in the presence of K2CO3 in an industrial methylated spirit to produce (S)-2-[l-[2-(2,3-benzofuran-5-yl)-2- oxoethyl]-3-pyrrolidinyl]-2,2-diphenylacetamide hydrochloride (VII), which is further hydrogenated in the presence of Pd/C in acetic acid to produce Darifenacin crude, followed by purification using column chromatography to produce pure Darifenacin (la), which is converted to Darifenacin hydrobromide (I) using aqueous hydrobromic acid in acetone.
The rocess is as shown in Scheme-Ill below:

The disadvantage with the above processes is the use of column chromatography in the purification of Darifenacin (la). Employing column chromatography technique is tedious and laborious and also involves use of large quantities of solvents, and hence is not suitable for industrial scale operations.
US 6,930,188 discloses a process for the preparation of Darifenacin hydrobromide (I), by condensing 2-(2,3-dihydrobenzofuran-5-yl)acetic acid (VIII) with (S)-2,2- diphenyl-2-(3-pyrrolidinyl)acetonitrile hydrobromide (IX) in the presence of carbonyldiimidazole in ethyl acetate to produce (S)-3-(cyanodiphenylmethyl)-l-[2- (2,3-dihydrobenzofuran-5-yl)acetyl]pyri lidine (X), which is further reduced in the presence of sodium borohydride and boron trifluoride tetrahydrofuran complex to produce (S)-2-{l-[2-(2,3-dihydrobenzofuran-5-yl)ethyl]-3-pyrrolidinyl}-2,2- diphenyl acetonitrile (XI), followed by treating with HBr to produce (S)-2-{ l-[2- (2,3-dihydrobenzofuran-5-yl)ethyl]-3-pyrrolidinyl}-2,2-diphenyl acetonitrile hydrobromide (XIa). Compound (XIa) is treated with potassium hydroxide at 50 to 60°C to produce Darifenacin (la), followed by treating with ion-exchange resin to produce Darifenacin toluene solvate (lb), which is further converted to Darifenacin hydrobromide (I) using 48% hydrobromic acid in 2-butanone.
The rocess is as shown in Scheme-IV below:

Darifenacin HBr
(I) It has now been found that, during the condensation of 5-(2-bromoethyl)-2,3- benzofuran (IV) with 3-(S)-(-)-(l -carbamoyl- l ,l-diphenylmethyl)pyrrolidine (III) to produce (S)-2-[l-[2-(2,3-benzofuran-5-yl)ethyl]-3-pyrroIidinyl]-2,2- diphenylacetamide (V), 3-(S)-(-)-(l -carbamoyl- l , l-diphenylmethyl)pyrrolidine (III) remained unreacted to about 8 to 10% in the reaction mass. It is difficult to separate the compound (III) through crystallization from Darifenacin hydrobromide (I), which typically require two to three crystallizations to achieve desired Darifenacin hydrobromide (I) purity. The second and third crystallization adds time to the manufacturing process and thus negatively impacts product throughput. Additionally, a second and third crystallization reduces yield as some Darifenacin hydrobromide (I) remains uncrystallized and is not recovered from the liquid phase.
Hence, there is a need to develop a purification process, which removes the unreacted intermediate compound 3-(S)-(-)-(l-carbamoyl-l ,l – diphenylmethyl)pyrrolidine (III) from the reaction mass, which in turn provides Darifenacin hydrobromide of high purity with improved yield.
Further, it has been found that Darifenacin produced by the condensation of 5-(2- bromoethyl)-2,3-dihydrobenzofuran (II) with 3-(S)-(-)-( 1 -carbamoyl- 1 ,1 – diphenylmethyOp rrolidine (III) contains dimmer impurity (XII).
Formula (XII)

Hence, there is a need to develop process, which reduces the unwanted Darifenacin dimer (XII), which is influenced by controlling the quantity of compound (XIII).

Formula (XIII)
PROCESS
(a) Dunn, P. J.; Matthews, J. G.; Newbury, T. J.; O’Connor, G.US 6,930,188 B2, 2005.
(b) Narayan, K; Reddy, J. M.; Rao, G.; Chary, S.; Islam, A.; SivakumaranWO 2011/D70419 A1, 2011.
(c) Evansa, P.; Thomas, J.; Davies, R. H.US 2003/0199494 A1, 2003.
(d) Bhanu, M. N.; Naik, S.; Bodkhe, A.; Soni, A.US 2011/0144354 A1, 2011.
(e) Merli, V.; Canavesi, A.; Baverio, P.US 7,442,806 B2, 2008.
(f) Merli, V.; Canavesi, A.; Baverio, P.US 2009/0156831 A1, 2009.
(G) WO2009125426A2.

PATENT
https://www.google.com/patents/WO2011070419A1?cl=en

EXAMPLE – 1
Stage-1:
PREPARATION OF 5-(2-TOSYLOXYETHYL)-2,3-
DIHYDROBENZOFURAN
2-(2,3-Dihydrobenzofuran-5yl)ethanol (65 g, 0.39 mol) was dissolved in dichloromethane (650 ml) at 20-25°C under nitrogen atmosphere. The solution was cooled to 0-5°C and p-toluenesulfonyl chloride (79.27 g, 0.41 mol) was added in one lot. Triethylamine (60.04 g, 0.59 mol) was added slowly at 0-10°C, stirred for ~ 15 h at 20-25°C and the reaction was monitored by HPLC. Water was added and stirred for 10 min at 20-25°C. Layers were separated and the aqueous layer was extracted with dichloromethane (130 ml). The organic layer was combined and washed with water (2 x 130 ml) at 20-25°C at pH 12 – 12.5. Finally the organic layer was washed with saturated brine solution (130ml) and concentrated to complete dryness under reduced pressure at 35-45°C. The product was crystallized from ethyl acetate and n- hexanes mixture.
Yield: 96.5 g
Chromatographic purity (By HPLC): 97.85%
Stage-2:
PREPARATION OF DARIFENACIN HYDROBROMIDE
3-(S)-(-)-(l -Carbamoyl- l , l -diphenylmethyl)pyrrolidine L-(+)-tartrate (10 g, 0.02 mol), anhydrous potassium carbonate (22.50 g, 0.16 mol) and 5-(2-tosyloxyethyl)- 2,3-dihydrobenzofuran (7 g, 0.02 mol) were suspended in anhydrous acetonitrile ( 100 ml) under nitrogen atmosphere at 25 ± 2°C. The reaction suspension was heated to 70 ± 2 °C and stirred for 4 h. Reaction progress was monitored by HPLC. The reaction mass was cooled to 30 + 2°C, the salts were filtered and washed with acetonitrile (10 ml). The filtrate was concentrated under reduced pressure at 50 ± 2 °C. The residue was dissolved in dichloromethane (50 ml), water (50 ml) was added and the pH was adjusted to 2 ± 0.1 with 24% w/w aqueous hydrobromic acid at 25- 30°C. The layers were separated and the aqueous layer was extracted with aqueous dichloromethane (20 ml). Water (50 ml) was added to the combined dichloromethane layer and pH was adjusted to 9 ± 0.1 with 25% w/w aqueous potassium carbonate solution at 25 ± 2°C. The layers were separated and concentrated under reduced pressure at 35-40°C. The residue was dissolved in acetone (50 ml), cooled to 5-10°C and the pH was adjusted to acidic with 48% w/w aqueous hydrobromic acid at 5-10°C. The residue was stirred for 2 h at 20-25°C, cooled to 0-5°C and stirred for 1 h at 0-5°C. The product was filtered, washed with chilled acetone (10 ml) and dried at 50-55°C.
Yield: 9.4 g
Chromatographic purity (By HPLC): 98.2%.
5 -(2-Tosy loxyethy l)-2, 3 -dihydrobenzofuran : Nil
Darifenacin dimer impurity: 0.96%.
EXAMPLE – 2
Stage-1 :
PREPARATION OF 5-(2-BROMOETHYL)-2,3-DIHYDROBENZOFURAN
2- (2,3-Dihydrobenzofuran-5-yl)ethanol (10 g, 0.06 mol) was dissolved in acetonitrile (60 ml) at 25 ± 2°C under nitrogen atmosphere and triphenylphosphine dibromide (27.02 g, 0.06 mol) was added in one lot at 25 ± 2°C. The reaction mass was heated to 76-78°C and stirred for 2 h. Reaction progress was monitored by TLC [Ethyl acetate: n-Hexanes; 2:8 v/v], Acetonitrile was completely distilled off under reduced pressure at 76-78°C. The residue was cooled and the product was extracted with n-hexanes (4 x 30 ml) at 25 ± 2°C. The solution was filtered and diluted with ethyl acetate (50 ml) and washed with 5% w/w aqueous sodium bicarbonate solution (2 x 50 ml) at 25 ± 2°C. The organic layer was concentrated under reduced pressure at 40-50°C.
Yield: 7 g
Stage-2
PREPARATION OF DARIFENACIN HYDROBROMIDE
3- (S)-(-)-(l -Carbamoyl- l ,l-diphenylmethyl)pyrrolidine L-(+)-tartrate (5 g, 0.01 mol), anhydrous potassium carbonate (1 1.25 g, 0.08 mol) and 5-(2-bromoethyl)-2,3- dihydrobenzofuran (2.5 g, 0.01 mol) were suspended in anhydrous acetonitrile (50 ml) under nitrogen atmosphere at 25 ± 2°C. The reaction suspension was heated to 70 ± 2 °C and stirred for 4 h. Reaction progress was monitored by HPLC. The reaction mass was cooled to 30 ± 2°C, salts were filtered and washed with acetonitrile (5 ml). The filtrate was concentrated under reduced pressure at 50 ± 2 0 C. The residue was dissolved in dichloromethane (25 ml), water (25 ml) was added and the pH was adjusted to 2 ± 0.1 with 24% w/w aqueous hydrobromic acid at 25- 30°C. The layers were separated and the aqueous layer was extracted with dichloromethane (10 ml). Water (25 ml) was added to the combined dichloromethane layer and pH was adjusted to 9 ± 0.1 with 25% w/w aqueous potassium carbonate solution at 25 ± 2°C. The layers were separated and the organic layer was concentrated under reduced pressure at 35-40°C. The residue was dissolved in acetone (25 ml), cooled to 5-10°C and the pH was adjusted to acidic with 48% w/w aqueous hydrobromic acid at 5-10°C. The residue was stirred for 2 h at 20-25°C, cooled to Q-5°C and stirred for 1 h at 0-5°C. The product was filtered, washed with chilled acetone (5 ml) and dried at 50-55°C.
Yield: 4.5 g
Chromatographic purity (By HPLC): 99.24%
5-(2-bromoethyl)-2,3-dihydiObenzofuran: Nil
Darifenacin dimer impurity: 0.39%.
EXAMPLE – 3
PURIFICATION OF DARIFENACIN HYDROBROMIDE Darifenacin hydrobromide (10 g) was suspended in acetic acid (15 g) at 25 ± 2°C and heated to 65-70°C. Activated carbon (0.25 g) was added and stin-ed for 15 min at 65-70°C. Carbon was filtered off through hyflo and washed with hot acetic acid (5 g). Water (200 ml) was added to the filtrate slowly at 50-55°C, cooled to 45°C and Darifenacin hydrobromide seed (0.05 g) was added. The resulting solution was cooled to 20-25 °C and stin-ed for 1 h and further cooled to 0-5 °C and stirred for 1 h. The solid was filtered and washed with cold water (10 ml). The product was dried at 50-55°C.
Yield: 7.6 g
Chromatographic purity (By HPLC): 99.52%
5-(2-bromoethyl)-2,3-dihydrobenzofuran: Nil 5-(2-Tosyloxyethyl)-253-dihydrobenzofuran: Nil
Darifenacin dimer impurity: 0.20%.
EXAMPLE – 4
PURIFICATION OF DARIFENACIN HYDROBROMIDE
Darifenacin hydrobromide (15 g) was suspended in a mixture of acetic acid (25 g) and water (25 ml) at 25 ± 2°C and heated to 65-70°C. Activated carbon (0.75 g) was added and stirred for 15 min at 65-70°C. Carbon was filtered off through hyflo and washed with a mixture of acetic acid and DM water (10 g). Water (120 ml) was added to the filtrate slowly at 50-55°C, cooled to 45°C and Darifenacin hydrobromide seed (0.05 g) was added. The resulting solution was cooled to 20- 25°C and stirred for 1 h and further cooled to 0-5°C and stirred for 1 h. The solid was filtered and washed with cold water (30 ml). The product was dried at 50-55°C. Yield: 1 1.9 g
Chromatographic purity (By HPLC): 99.71 %
5-(2-bromoethyl)-2,3-dihydrobenzofuran: Nil
5-(2-Tosyloxyethyl)-2,3-dihydrobenzofuran: Nil
Darifenacin dimer impurity: 0.20%. EXAMPLE – 5
PURIFICATION OF DARIFENACIN HYDROBROMIDE
Darifenacin hydrobromide (9 g) was suspended in acetone (45 ml) at 25 ± 2°C, heated to 55-60°C and stirred for 30 + 5 min at 55-60°C. The resulting solution was cooled to 20-25°C and stin-ed for 30 + 5 min, which is further cooled to 0-5°C and stirred for 1 h. The solid was filtered and washed with chilled acetone (9 ml). The product was dried at 50-55°C.
Yield: 8.8 g
Chromatographic purity (By HPLC): 99.87%
5-(2-bromoethyl)-2,3-dihydrobenzofuran: Nil
5-(2-Tosyloxyethyl)-2,3-dihydrobenzofuran: Nil
Darifenacin dimer impurity: 0.08%. EXAMPLE – 6
PURIFICATION OF DARIFENACIN HYDROBROMIDE
Darifenacin hydrobromide (9 g) was suspended in a mixture of acetone (45 ml) and DM water (1.77 ml) at 25 ± 2°C, heated to 55-60°C and stirred for 30 + 5 min at 55- 60°C. The resulting solution was cooled to 20-25°C and stirred for 30 + 5 min, which was further cooled to 0-5°C and stirred for 1 h. The product was filtered and washed with chilled acetone (9 ml). The product was dried at 50-55°C.
Yield: 8.4 g
Chromatographic purity (By HPLC): 99.88%
EXAMPLE – 7
PURIFICATION OF DARIFENACIN HYDROBROMIDE
Darifenacin hydrobromide (10 g) was suspended in a mixture of acetone (50 ml) and DM water (3.95 ml) at 25 ± 2°C, heated to 55-58°C and stirred for 30 ± 5 min. The resulting solution was cooled to 20-25°C and stirred for 30 ± 5 min, which was further cooled to 0-5 °C and stirred for 1 hour. The product was filtered and washed with chilled acetone (10ml, 0-5°C). The product was dried at 50-55°C.
Yield: 8.30g
Chromatographic Purity (By HPLC): 99.83 %
Darifenacin dimmer: 0.10%
EXAMPLE – 8
PURIFICATION OF DARIFENACIN HYDROBROMIDE
Darifenacin hydrobromide (10 g) was suspended in a mixture of acetone (50 ml) and DM water (7.9 ml) at 25 ± 2°C, heated to 55-60°C and stirred for 30 + 5 min. The resulting solution was cooled to 20-25°C and stirred for 30 ± 5 min, which was further cooled to 0-5°C and stirred for 1 hour. The product was filtered and washed with chilled acetone (10 ml, 0-5°C). The product was dried at 50-55°C.
Yield: 6.70g
Chromatographic Purity (By HPLC): 99.94 %
Darifenacin dimmer: Nil.
Paper
A New Solvent System (Cyclopentyl Methyl Ether–Water) in Process Development of Darifenacin HBr

Darifenacin is a potent and competitive M3 selective receptor antagonist (M3SRA), and its hydrobromide salt (1) is the active ingredient of pharmaceutical formulations for oral treatment of urinary incontinence. The present work demonstrates an efficient, commercial manufacturing process for darifenacin hydrobromide (1).
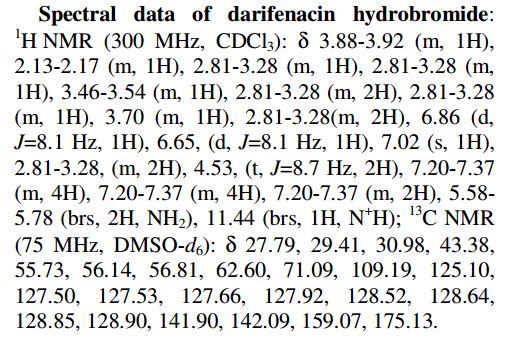
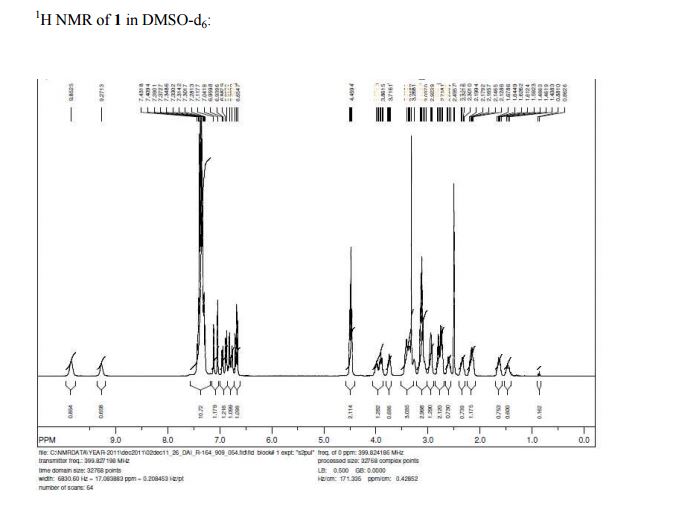
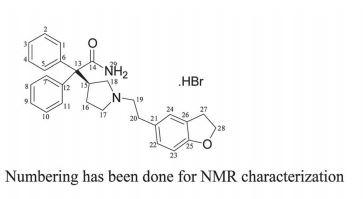
1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.8 (bs, 0.7H), 9.3 (bs, 0.3 H), 7.4–7.3 (m, 10 H), 7.1–7.0 (m, 1H), 7.0–6.7 (m, 2H), 6.7 (m, 1H), 4.5 (m, 2H), 4.0–3.9 (m, 1.3 H), 3.8–3.7 (m, 0.7 H), 3.4–3.3 (m, 2H), 3.1 (m, 2H), 2.9 (m, 1.3 H), 2.8–2.7 (m, 2H), 2.6 (m, 0.7H), 2.4–2.3 (m, 0.7H), 2.2 (m, 1.3H), 1.6 (m, 0.7 H), 1.5 (m, 0.3 H).
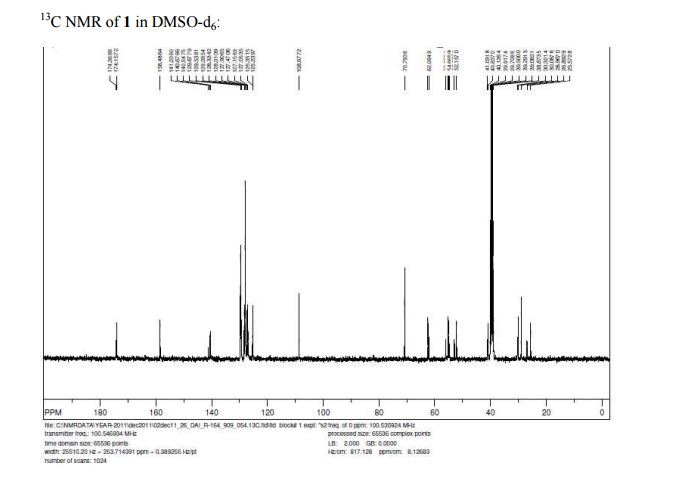
13C NMR (DMSO-d6, 100 MHz, δ ppm): 174.4, 174.2, 158.5, 141.2, 140.7, 140.6, 129.7, 129.4, 129.5, 128.3, 128.0, 127.9, 127.5, 127.2, 127.1, 125.4, 125.2, 108.7, 70.8, 62.4, 62.1, 56.1, 55.2, 55.1, 54.7, 53.0, 52.2, 40.0, 40.8, 30.3, 30.1, 29.0, 26.9, 25.6.
Calcd for C28H30N2O2·HBr, (M+)/z: 425.56; found (M + H)/z 427.2, (M + Na)/z 449.3.
Anal. Calcd for C28H31BrN2O2: C, 66.27; H, 6.16; N, 5.52. Found: C, 66.36; H, 6.07; N, 5.68.
PATENT
https://www.google.com/patents/WO2009094957A1?cl=en
Scheme 4:

Example 1

Advanced intermediate VII (4.3 g; 0.01 mol) is stirred up in an aqueous solution of potassium phosphate (9.43 g; 0.041 mol in 20 ml of water) at the laboratory temperature. A toluene solution (20 ml) of intermediate VIII (2.41 g; 0.011 mol) is added to the mixture and the mixture is heated up in an oil bath T=90 0C while being stirred for 3.5 h. After cooling the toluene layer is separated and the aqueous layer is extracted with toluene. The combined toluene extracts are shaken with water and the solvent is distilled off at a reduced pressure. The evaporation residue is dissolved in ethylmethylketone, and an equimolar amount of 48% hydrobromic acid is added. The separated darifenacin hydrobromide is filtered off and dried.
Yield: 85% of theory.
Example 2

Advanced intermediate VII (4.3 g; 0.01 mol) is stirred up in an aqueous solution of potassium carbonate (6.1 g; 0.044 mol in 20 ml of water) at the laboratory temperature. A toluene solution (20 ml) of intermediate VIII (2.41 g; 0.011 mol) is added to the mixture and the mixture is heated in an oil bath T=90 °C while being stirred for 3.5 h. After cooling the toluene layer is separated and the aqueous layer is extracted with toluene. The combined toluene extracts are shaken with water and the solvent is distilled off at a reduced pressure. The evaporation residue is dissolved in ethylmethylketone, and an equimolar amount of 48% hydrobromic acid is added. The separated darifenacine hydrobromide is filtered off and dried.
Yield: 86% of theory.
Example 3

Advanced intermediate VII (4.3 g; 0.01 mol) is stirred up in an aqueous solution of potassium phosphate (9.43 g; 0.041 mol in 20 ml of water) at the laboratory temperature. A solution of intermediate VIII (2.41 g; 0.011 mol) in cyclohexane (20 ml) is added to the mixture and the mixture is heated in an oil bath T=90 0C while being stirred for 3.5 h. The layers are separated while hot. The cyclohexane solution is cooled to the laboratory temperature under intensive stirring. This way the darifenacin base is separated. The product is filtered off and dried. The base is dissolved in ethylmethylketone, and an equimolar amount of 48% hydrobromic acid is added. The separated darifenacin hydrobromide is filtered off and dried.
Yield: 85% of theory.
clip

References
External links
- Enablex product website, run by Warner Chilcott
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2011070419A1 * | Dec 3, 2010 | Jun 16, 2011 | Aurobindo Pharma Limited | An improved process for the preparation of darifenacin hydrobromide |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2003080599A1 | Mar 17, 2003 | Oct 2, 2003 | Novartis International Pharmaceutical Ltd. | Stable hydrate of a muscarinic receptor antagonist |
| WO2007076157A2 * | Dec 27, 2006 | Jul 5, 2007 | Teva Pharmaceuticals Industries Ltd. | Processes for preparing darifenacin hydrobromide |
| WO2007076158A2 * | Dec 27, 2006 | Jul 5, 2007 | Teva Pharmaceutical Industries Ltd. | Processes for preparing darifenacin hydrobromide |
| WO2007076159A2 | Dec 27, 2006 | Jul 5, 2007 | Teva Pharmaceutical Industries Ltd. | Pure darifenacin hydrobromide substantially free of oxidized darifenacin and salts thereof and processes for the preparation thereof |
| EP0388054A1 | Mar 2, 1990 | Sep 19, 1990 | Pfizer Limited | Pyrrolidine derivatives |
| WO2009094957A1 * | Jan 14, 2009 | Aug 6, 2009 | Zentiva, K.S. | A method for the preparation of darifenacin hydrogen bromide |
| US5096890 | Mar 13, 1990 | Mar 17, 1992 | Pfizer Inc. | Pyrrolidine derivatives |
| US6930188 | Mar 25, 2003 | Aug 16, 2005 | Novartis International Pharmaceutical, Ltd. | Stable hydrate of a muscarinic receptor antagonist |
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Enablex |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a605039 |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | G04BD10 (WHO) |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 15 to 19% (dose-dependent) |
| Protein binding | 98% |
| Metabolism | Hepatic (CYP2D6– and CYP3A4-mediated) |
| Biological half-life | 13 to 19 hours |
| Excretion | Renal (60%) and biliary (40%) |
| Identifiers | |
| CAS Number | 133099-04-4 |
| PubChem (CID) | 444031 |
| IUPHAR/BPS | 321 |
| DrugBank | DB00496 |
| ChemSpider | 392054 |
| UNII | APG9819VLM |
| KEGG | D01699 |
| ChEBI | CHEBI:391960 |
| ChEMBL | CHEMBL1346 |
| ECHA InfoCard | 100.118.382 |
| Chemical and physical data | |
| Formula | C28H30N2O2 |
| Molar mass | 426.55 g/mol |
| 3D model (Jmol) | Interactive image |
/////////Darifenacin, 臭化水素酸ダリフェナシン , Antispasmodic, Antimuscarinic, UK-88525-04, Emselex® , Enablex® , Xelena®,
C1CN(CC1C(C2=CC=CC=C2)(C3=CC=CC=C3)C(=O)N)CCC4=CC5=C(C=C4)OCC5