














HEC-68498, CT-365
CAS 1621718-37-3
N-[5-(3-Cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxy-3-pyridinyl]-2,4-difluorobenzenesulfonamide
HEC Pharm , Calitor Sciences Llc; Sunshine Lake Pharma Co Ltd
PHASE 1, idiopathic pulmonary fibrosis and solid tumors
Phosphoinositide 3-kinase inhibitor; mTOR inhibitor

- Originator HEC Pharm
- Developer HEC Pharm; Sunshine Lake Pharma
- Class Anti-inflammatories; Antifibrotics; Isoenzymes
- Mechanism of Action 1 Phosphatidylinositol 3 kinase inhibitors; MTOR protein inhibitors
- Phase I Idiopathic pulmonary fibrosis
- 22 May 2018 Phase-I clinical trials in Idiopathic pulmonary fibrosis in USA (PO) (NCT03502902)
- 24 Apr 2018 Sunshine Lake Pharma in collaboration with Covance plans a phase I trial for Idiopathic pulmonary fibrosis (In volunteers) in China , (NCT03502902)
- 19 Apr 2018 Preclinical trials in Idiopathic pulmonary fibrosis in China (PO)
- US 20140234254
- CN 103965199
CN 103965199
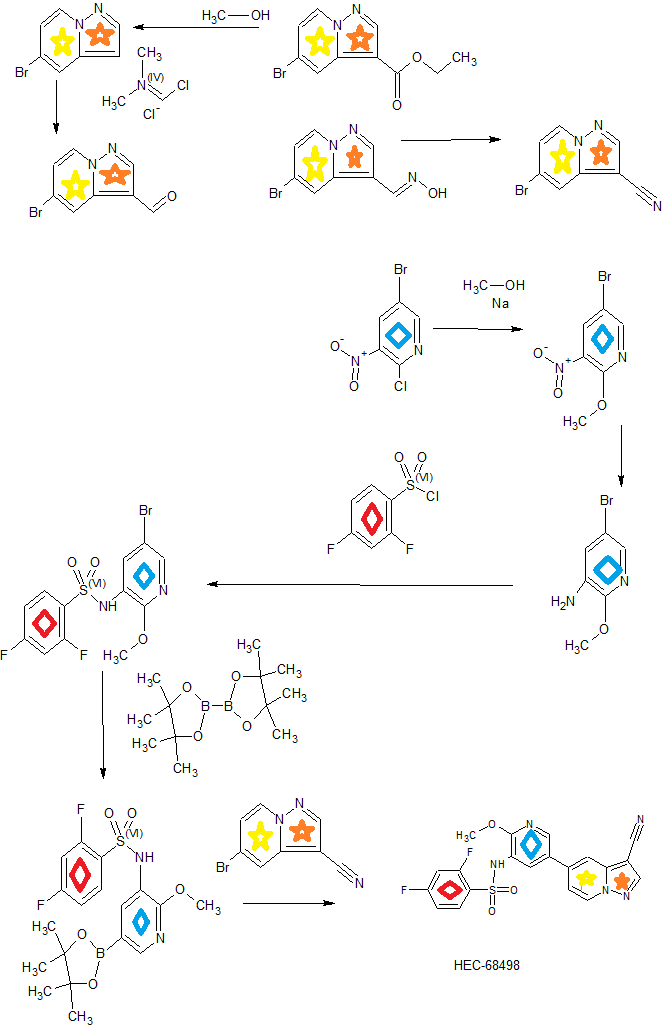
CN 103965199
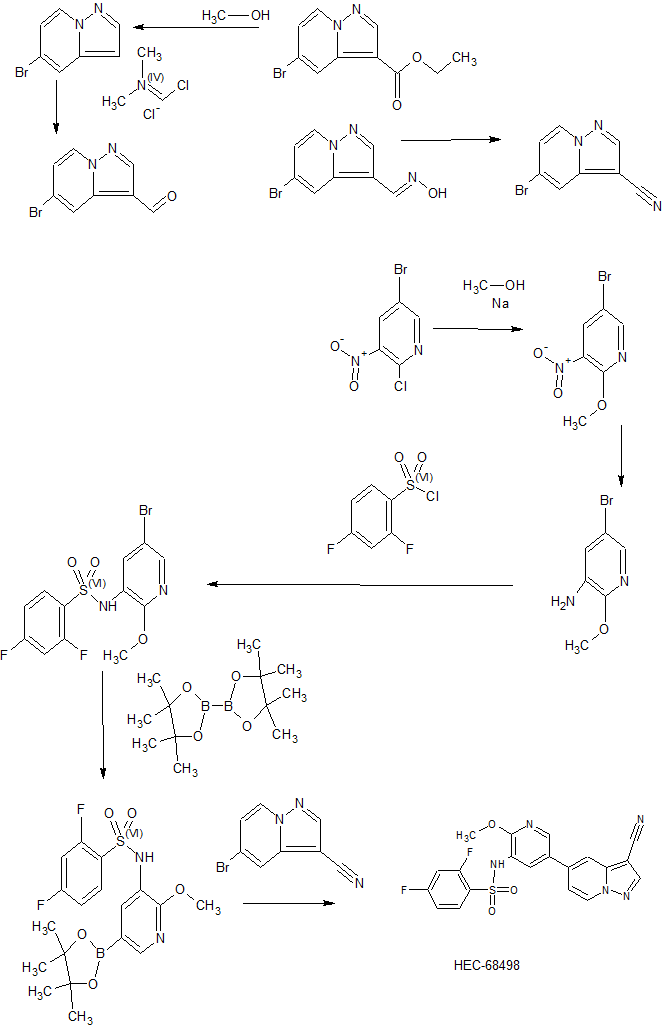
Sunshine Lake Pharma , a subsidiary of HEC Pharm is developing an oral capsule formulation of HEC-68498 (phase 1, in July 2019) sodium salt, a dual inhibitor of phosphoinositide-3 kinase and the mTOR pathway, for the treatment of idiopathic pulmonary fibrosis and solid tumors
HEC 68498 is an oral inhibitor of phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin in clinical development at HEC Pharm for the treatment of idiopathic pulmonary fibrosis. A phase I trial is under way in healthy volunteers.
The phosphoinositide 3-kinases (PI3 kinases or PI3Ks), a family of lipid kinases, have been found to play key regulatory roles in many cellular processes including cell survival, proliferation and differentiation. The PI3K enzymes consist of three classes with variable primary structure, function and substrate specificity. Class I PI3Ks consist of heterodimers of regulatory and catalytic subunits, and are subdivided into 1A and 1B based on their mode of activation. Class 1A PI3Ks are activated by various cell surface tyrosine kinases, and consist of the catalytic pl lO and regulatory p85 subunits. The three known isoforms of Class 1A pl lO are pl lOot, rΐ ΐqb, and rΐ ΐqd, which all contain an amino terminal regulatory interacting region (which interfaces with p85), a Ras binding domain, and a carboxy terminal catalytic domain. Class IB PI3Ks consist of the catalytic (pl lOy) and regulatory (p 101 ) subunits and are activated by G-protein coupled receptors. (“Small-molecule inhibitors of the PI3K signaling network” Future Med. Chem ., 2011, 3, 5, 549-565).
[0004] As major effectors downstream of receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs), PI3Ks transduce signals from various growth factors and cytokines into intracellular massages by generating phospholipids, which activate the serine-threonine protein kinase ART (also known as protein kinase B (PKB)) and other downstream effector pathways. The tumor suppressor or PTEN (phosphatase and tensin
homologue) is the most important negative regulator of the PI3K signaling pathway. (“Status of PBK/Akt/mTOR Pathway Inhibitors in Lymphoma.” Clin Lymphoma, Myeloma Leuk , 2014, 14(5), 335-342.)
[0005] The signaling network defined by phosphoinositide 3-kinases (PI3Ks), AKT and mammalian target of rapamycin (mTOR) controls most hallmarks of cancer, including cell cycle, survival, metabolism, motility and genomic instability. The pathway also contributes to cancer promoting aspects of the tumor environment, such as angiogenesis and inflammatory cell recruitment. The lipid second messenger produced by PI3K enzymes, phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3; also known as PIP3), is constitutively elevated in most cancer cells and recruits cytoplasmic proteins to membrane-localized‘onco’ signal osomes.
[0006] Cancer genetic studies suggest that the PI3K pathway is the most frequently altered pathway in human tumors: the PIK3CA gene (encoding the PI3K catalytic isoform pl lOa) is the second most frequently mutated oncogene, and PTEN (encoding phosphatase and tensin homolog, the major PtdIns(3,4,5)P3 phosphatase) is among the most frequently mutated tumor suppressor genes. In accord, a recent genomic study of head and neck cancer found the PI3K pathway to be the most frequently mutated. Indeed, even in cancer cells expressing normal PI3K and PTEN genes, other lesions are present that activate the PI3K signaling network (that is, activated tyrosine kinases, RAS and AKT, etc ). As a net result of these anomalies, the PI3K pathway is activated, mutated or amplified in many malignancies, including in ovarian cancer (Campbell et al., Cancer Res., 2004, 64, 7678-7681; Levine et al., Clin. Cancer Res., 2005, 11, 2875-2878; Wang et al., Hum. Mutat., 2005, 25, 322; Lee et al., Gynecol. Oncol. ,2005, 97, 26-34), cervical cancer, breast cancer (Bachman et al.,· Cancer Biol., Ther, 2004, 3, 772-775; Levine et al., supra; Li et al., Breast Cancer Res. Treat., 2006, 96, 91-95; Saal et al., Cancer Res., 2005, 65, 2554-2559; Samuels and Velculescu, Cell Cycle, 2004, 3, 1221-1224), colorectal cancer (Samuels et al., Science, 2004, 304, 554; Velho et al., Eur. J. Cancer, 2005, 41, 1649-1654), endometrial cancer (Oda et al ., Cancer Res., 2005, 65, 10669-10673), gastric carcinomas (Byun et al., M. J. Cancer, 2003 , 104, 318-327; Li et al., supra; Velho et al., supra; Lee et al., Oncogene, 2005 , 24, 1477-1480), hepatocellular carcinoma (Lee et al., id), small and non-small cell lung cancer (Tang et al., Lung Cancer 2006, 11, 181-191; Massion et al , Am. J. Respir. Crit. Care Med., 2004, 170, 1088-1094), thyroid carcinoma (Wu et al., J. Clin. Endocrinol. Metab., 2005, 90, 4688-4693),
acute myelogenous leukemia (AML) (Sujobert et al., Blood, 1997, 106, 1063-1066), chronic myelogenous leukemia (CML) (Hickey et al., J. Biol. Chem ., 2006, 281, 2441-2450), glioblastomas (Hartmann et al. Jlcta Neuropathol (Bert ), 2005, 109, 639-642; Samuels et al., supra), Hodgkin and non-Hodgkin lymphomas (“PI3K and cancer: lessons, challenges and opportunities”, Nature Reviews Drug Discovery., 2014, 13, 140).
[0007] The PI3K pathway is hyperactivated in most cancers, yet the capacity of PI3K inhibitors to induce tumor cell death is limited. The efficacy of PI3K inhibition can also derive from interference with the cancer cells’ ability to respond to stromal signals, as illustrated by the approved PI3K5 inhibitor idelalisib in B-cell malignancies. Inhibition of the leukocyte-enriched PI3K5 or RI3Kg may unleash antitumor T-cell responses by inhibiting regulatory T cells and immune-suppressive myeloid cells. Moreover, tumor angiogenesis may be targeted by PI3K inhibitors to enhance cancer therapy. (“Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy”, Cancer Discov., 2016, 6(10), 1090-1105.)
[0008] mTOR is a highly conserved serine-threonine kinase with lipid kinase activity and participitates as an effector in the PI3K/AKT pathway. mTOR exists in two distinct complexes, mTORCl and mTORC2, and plays an important role in cell proliferation by monitoring nutrient avaliability and cellular energy levels. The downstream targets of mTORCl are ribosomal protein S6 kinase 1 and eukaryotic translation initiation factor 4E-binding protein 1, both of which are crucial to the regulation of protein synthesis. (“Present and future of PI3K pathway inhibition in cancer: perspectives and limitations”, Current Med. Chem., 2011, 18, 2647-2685).
[0009] Knowledge about consequences of dysregulated mTOR signaling for tumorigenesis comes mostly from studies of pharmacologically disruption of mTOR by repamycin and its analogues such as temsirolimus (CCI-779) and everolimus (RADOOl).Rapamycin was found to inhibit mTOR and thereby induce G1 arrest and apoptosis. The mechanism of rapamycin growth inhibition was found to be related to formation of complexes of rapamycin with FK-binding protein 12 (FKBP-12). These complexes then bound with high affinity to mTOR, preventing activation and resulting in inhibition of protein translation and cell growth. Cellular effects of mTOR inhibition are even more pronounced in cells that have concomitant inactivation of PTEN. Antitumor activity of rapamycin was subsequently identified, and a number of rapamycin analogues such as temsirolimus and everolimus have been approved by the US Food and Drug
Administration for the treatment certain types of cancer.
[0010] Fibrosis is the formation of excess fibrous connective tissue in an organ or tissue in a reparative or reactive process. Examples of fibrosis include, but are not limited to pulmonary fibrosis, liver fibrosis, dermal fibrosis, and renal fibrosis. Pulmonary fibrosis, also called idiopathic pulmonary fibrosis (IPF), interstitial diffuse pulmonary fibrosis, inflammatory pulmonary fibrosis, or fibrosing alveolitis, is a lung disorder and a heterogeneous group of conditions characterized by abnormal formation of fibrous tissue between alveoli caused by alveolitis comprising cellular infiltration into the alveolar septae with resulting fibrosis. The effects of IPF are chronic, progressive, and often fatal.
[0011] The clinical course of IPF is variable and largely unpredictable. IPF is ultimately fatal, with historical data suggesting a median survival time of 2-3 years from diagnosis. A decline in forced vital capacity (FVC) is indicative of disease progression in patients with IPF and change in FVC is the most commonly used endpoint in clinical trials. A decline in FVC of 5% or 10% of the predicted value over 6-12 months has been associated with increased mortality in patients with IPF.
[0012] Our understanding of the pathogenesis of IPF has evolved from that of a predominantly inflammatory disease to one driven by a complex interplay of repeated epithelial cell damage and aberrant wound healing, involving fibroblast recruitment, proliferation and differentiation, and culminating in excess deposition of extracellular matrix. This shift in knowledge prompted a change in the type of compounds being investigated as potential therapies, with those targeted at specific pathways in the development and progression of fibrosis becoming the focus.
[0013] In patients with IPF, the mechanisms whereby PI3K/mTOR inhibitors act may involve inhibition of kinases such as PI3Ks and mTOR. This results in inactivation of cellular receptors for mediators involved in the development of pulmonary fibrosis. As a result, fibroblast proliferation is inhibited and extracellular matrix deposition is reduced. (“Update on diagnosis and treatment of idiopathic pulmonary fibrosis”, J Bras Pneumol. 2015, 41(5), 454-466.)
[0014] Accordingly, small-molecule compounds that specially inhibit, regulate and/or modulate the signal transduction of kinases, particularly including PI3K and mTOR as described above, are desirable as a means to prevent, manage, or treat proliferative disorders and fibrosis, particular idiopathic pulmonary fibrosis in a patient. One such small-molecule is A-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfon-amide, which has the chemical structure as shown in the following:
[0015] WO 2014130375A1 described the synthesis of N-(5 -(‘3 -cyanopyrazol o [l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (Example 3) and also disclosed the therapeutic activity of this molecule in inhibiting, regulating and modulating the signal transduction of protein kinases.
[0016] Different salts and solid state forms of an active pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid state forms may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, improving the dissolution profile, stability (polymorph as well as chemical stability) and shelf-life. These variations in the properties of different salts and solid state forms may also provide improvements to the final dosage form, for example, if they serve to improve bioavailability. Different salts and solid state forms of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which may in turn provide additional opportunities to assess variations in the properties and characteristics of a solid active pharmaceutical ingredient.
Different salts and solid state forms of /V-(5-(3-cyanopyrazolo[l,5- ]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide are described herein.
PATENT
WO2014130375 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014130375
claiming new pyrazolo[1,5-a]pyridine derivatives are PI3K and mTOR inhibitors, useful for treating proliferative diseases
Example 3 N-(5-(3-cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide
Step 1) 5-bromopyrazolo[1,5-a]pyridine
[196] A solution of ethyl 5-bromopyrazolo[1,5-a]pyridine-3-carboxylate (240
mmol) in 40% H2SO4 (12 mL) was stirred at 100 °C for 4 hours, then cooled to rt, and neutralized to pH=7 with aq. NaOH (6 M) in ice bath. The resulted mixture was extracted with DCM (25 mL x 2). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo to give the title compound as a light yellow solid (175 mg, 99.5%).
MS (ESI, pos. ion) m/z: 196.9 [M+H]+.
Step 2) 5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde
[197] To a solution of 5-bromopyrazolo[1,5-a]pyridine (175 mg, 0.89 mmol) in DCM (6 mL) was added (chloromethylene)dimethyliminium chloride (632 mg, 3.56 mmol). The reaction was stirred at 44 °C overnight, and concentrated in vacuo. The residue was dissolved in saturated NaHCO3 aqueous solution (25 mL) and the resulted mixture was then extracted with EtOAc (25 mL x 3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo to give the title compound as a light yellow solid (225 mg, 100%).
MS (ESI, pos. ion) m/z: 225.0 [M+H]+.
Step 3) (E)-5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde oxime
[198] To a suspension of 5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde (225 mg, 1 mmol) in EtOH (10 mL) and H2O (5 mL) was added hydroxylamine hydrochloride (104 mg, 1.5 mmol). The reaction was stirred at 85 °C for 2 hours, then cooled to rt, and concentrated in vacuo. The residue was adjusted to pH=7 with saturated NaHCO3 aqueous solution. The resulted mixture was then filtered and the filter cake was dried in vacuo to give title compound as a yellow solid (240 mg, 99%).
MS (ESI, pos. ion) m/z: 240.0 [M+H]+.
Step 4) 5-bromopyrazolo[1,5-a]pyridine-3-carbonitrile
[199] A solution of (E)-5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde oxime (240 mg,
1 mmol) in Ac2O (6 mL) was stirred at 140 °C for 18 hours, then cooled to rt, and concentrated in vacuo. The residue was washed with Et2O (1 mL) to give the title compound as a yellow solid (44 mg, 22.5%).
MS (ESI, pos. ion) m/z: 222.0 [M+H]+.
Step 5) N-(5-(3-cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide
[200] 2,4-difluoro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)benzenesulfonamide (612 mg, 1.5 mmol), 5-bromopyrazolo[1,5-a]pyridine-3-carbonitrile (222 mg, 1 mmol), Pd(dppf)Cl2·CH2Cl2 (16 mg, 0.02 mmol) and Na2CO3 (85 mg, 0.8 mmol) were placed into a two-neck flask, then degassed with N2 for 3 times, and followed by adding 1,4-dioxane (5 mL) and water (1 mL). The resulted mixture was degassed with N2 for 3 times, then heated to 90 °C and stirred further for 5 hours. The mixture was cooled to rt and filtered. The filtrate was concentrated in vacuo and the residue was purified by a silica gel column chromatography (PE/EtOAc (v/v) = 1/2) to give the title compound as a light yellow solid (400 mg, 81.6%).
MS (ESI, pos. ion) m/z: 442.0 [M+H]+;
1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.37 (s, 1H), 9.02 (d, J = 7.2 Hz, 1H), 8.67 (s, 1H), 8.60 (d, J = 2.2 Hz, 1H), 8.26-8.16 (m, 2H), 7.82-7.72 (m, 1H), 7.57 (dd, J = 13.2, 5.8 Hz, 2H), 7.21 (t, J= 8.5 Hz, 1H), 3.67 (s, 3H).
PATENT
WO-2019125967
The invention relates to salts of pyrazolo[l,5-a]pyridine derivatives and use thereof, specifically relates to salt of /V-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl) -2,4-difluorobenzenesulfonamide (compound of formula (I)) and use thereof, further relates to composition containing said salts above. The salts or the composition can be used to inhibit/modulate protein kinases, further prevent, manage or treat proliferative disorders or pulmonary fibrosis in a patient.
Amorphous form of mono-sodium salt of HEC-68498 , useful for treating a proliferative disorder or pulmonary fibrosis.
The invention is further illustrated by the following examples, which are not be construed as limiting the invention in scope.
[00108] /V-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluoroben zenesulfonamide can be prepared according to the synthetic method of example 3 disclosed in WO2014130375 Al.
//////////////HEC-68498, HEC 68498, HEC68498, HEC Pharm , Calitor Sciences, Sunshine Lake Pharma, PHASE 1, proliferative disorder, pulmonary fibrosis, idiopathic pulmonary fibrosis, solid tumors, CT-365 , CT 365 , CT365
Fc1ccc(c(F)c1)S(=O)(=O)Nc2cc(cnc2OC)c3ccn4ncc(C#N)c4c3