
LASMIDITAN, COL-144 , LY-573144
- Molecular FormulaC19H18F3N3O2
- Average mass377.360 Da
-
ласмидитан
لاسميديتان
613677-28-4 HYDROCHLORIDE
439239-90-4 (free base)
SUCCINATE 439239-92-6
2,4,6-Trifluoro-N-[6-(1-methylpiperidin-4-ylcarbonyl)pyridin-2-yl]benzamide
2,4,6-trifluoro-N-{6-[(1-methylpiperidin-4-yl)carbonyl]pyridin-2-yl}benzamide
CoLucid Pharmaceuticals, PHASE 3, MIGRAINE
UNII:760I9WM792
Lasmiditan is an oral medication used in the termination of migraine headaches that was first approved for use in the United States in October 2019.
A high-affinity, highly selective serotonin 5-HT(1F) receptor agonist.
Lasmiditan, also known as COL-144 and LY573144, is a novel, centrally acting, highly selective 5-HT(1F) receptor agonist (K1=2.21 μM) without vasoconstrictor activity that seemed effective when given as an intravenous infusion in a proof-of-concept migraine study. Lasmiditan showed efficacy in its primary endpoint, with a 2-hour placebo-subtracted headache response of 28.8%, though with frequent reports of dizziness, paresthesias, and vertigo.
Product Ingredients
| Lasmiditan succinate; UNII-W64YBJ346B; Lasmiditan succinate [USAN]; W64YBJ346B; 439239-92-6; Lasmiditan succinate (USAN)
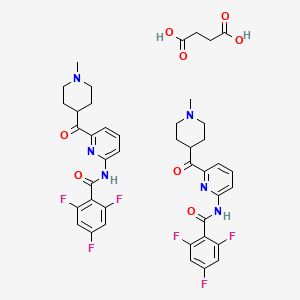
|
| Molecular Formula: |
C42H42F6N6O8 |
| Molecular Weight: |
872.822 g/mol |

Patent and Exclusivity for: N211280
Patent Data
Exclusivity Data
Lasmiditan, sold under the brand name Reyvow, is a medication used for the acute (active but short-term) treatment of migraine with or without aura (a sensory phenomenon or visual disturbance) in adults.[2] It is not useful for prevention.[2] It is taken by mouth.[2]
Common side effects include sleepiness, dizziness, tiredness, and numbness.[3][4]
Lasmiditan was approved in the United States in October 2019[3] and became available in February 2020.[5] It was developed by Eli Lilly.[3] The U.S. Food and Drug Administration (FDA) considers it to be a first-in-class medication.[6]
WO-2018010345, from Solipharma and the inventor on this API. Eli Lilly , following its acquisition of CoLucid Pharmaceuticals , is developing lasmiditan, a 5-HT 1f agonist, for treating acute migraine.
SYNTHESIS
SYN1
Synthetic Reference
Sheng, Xiaohong; Sheng, Xiaoxia; Jiang, Xiawei. Preparation of crystalline form of lasmiditan and its pharmaceutical composition. Assignee SoliPharma LLC, Peop. Rep. China. WO 2018010345. (2018).

SYN2
Synthetic Description
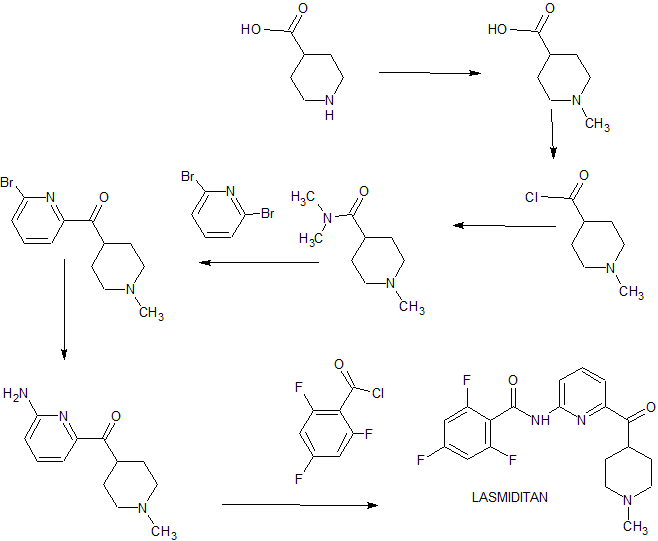
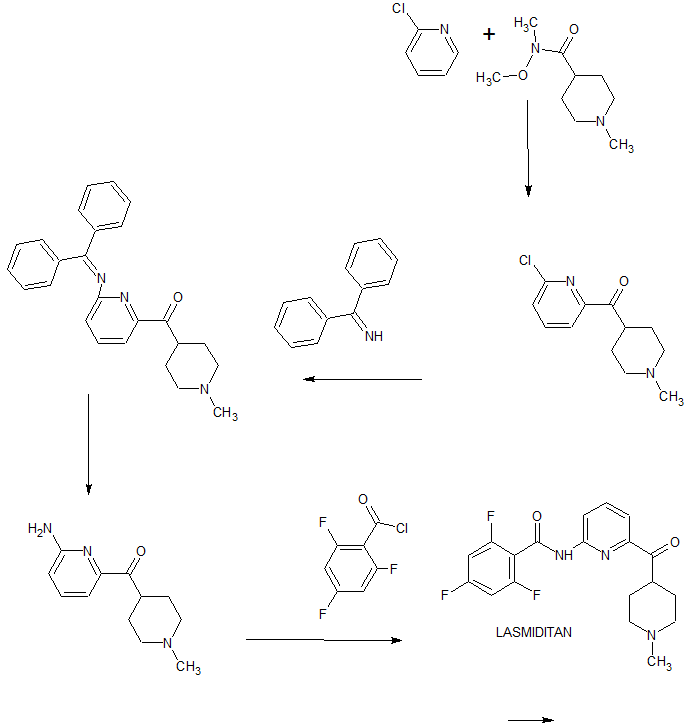
Reference: Carniaux, Jean-Francois; Cummins, Jonathan. Compositions and methods of synthesis of pyridinoylpiperidine derivatives as 5-HT1F agonists for treating and preventing migraine. Assignee Colucid Pharmaceuticals, Inc., USA. WO 2011123654. (2011).

SYN3
Synthetic Description
Reference: Cohen, Michael Philip; Kohlman, Daniel Timothy; Liang, Sidney Xi; Mancuso, Vincent; Victor, Frantz; Xu, Yao-Chang; Ying, Bai-Ping; Zacherl, Deanna Piatt; Zhang, Deyi. Preparation of pyridinoylpiperidines as 5-HT1F agonists. Assignee Eli Lilly and Company, USA. WO 2003084949. (2003).

SYN
SYN 2
REF
https://www.sciencedirect.com/science/article/abs/pii/S0223523420306395
5 Lasmiditan (Reyvow). Lasmiditan, developed by Eli Lilly, is a highly selective
agonist of 5-HT1F receptors [87]. The FDA approved lasmiditan as the first
neutrally-acting medication to treat migraine headaches [88]. Lasmiditan is a selective
5-HT1F agonist, but inactive against other 5-HT receptors or monoamine receptors
[89,90]. Unlike the triptan class of anti-migraine medications that lead to blood
pressurelability and other cardiovascular side effects, lasmiditan could terminate
migraines but without vasoconstriction [91]. However, lasmiditan may cause
significant driving impairment due to the CNS depression. And it is not a preventive
medication for migraine [92].
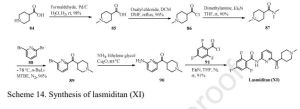
Eli Lilly has disclosed a kilogram-scale procedure to lasmiditan, which is described in
Scheme 14 [93]. Borch reduction of piperidine-4-carboxylic acid 84 gave 85. Further
chlorination with oxalyl chloride formed acyl chloride 86 in good yield. Next,
substitution with commercial dimethylamine, followed by coupling with
2,6-dibromopyridine 88 with assistance of tert-butyllithium gave rise to coupling
product 89 in 84% yield within the two-step sequence. Copper-catalyzed amination
between 89 and NH3 in ethylene glycol gave aminate product 90, which then treated
with acyl chloride 91 to give lasmiditan (XI) in 91% yield
CLICK ON IMAGE TO EXPAND
[87] T.V. Dupre, D.P. Jenkins, R.C. Muise-Helmericks, R.G. Schnellmann, The
5-hydroxytryptamine receptor 1F stimulates mitochondrial biogenesis and
angiogenesis in endothelial cells, Biochem. Pharmacol. 169 (2019) 113644.
[88] Y.N. Lamb, Lasmiditan: first approval, Drugs 79 (2019) 1989-1996.
[89] E. Rubio-Beltran, A. Labastida-Ramirez, K.A. Haanes, A. van den Bogaerdt, A.
Bogers, E. Zanelli, L. Meeus, A.H.J. Danser, M.R. Gralinski, P.B. Senese, K.W.
Johnson, J. Kovalchin, C.M. Villalon, A. MaassenVanDenBrink,
Characterization of binding, functional activity, and contractile responses of the
selective 5-HT1F receptor agonist lasmiditan, Br. J. Pharmacol. 176 (2019)
4681-4695.
[90] G.M. Dubowchik, C.M. Conway, A.W. Xin, Blocking the CGRP pathway for
acute and preventive treatment of migraine: the evolution of success, J. Med.
Chem. 63 (2020) 6600–6623.
[91] B. Kuca, S.D. Silberstein, L. Wietecha, P.H. Berg, G. Dozier, R.B. Lipton, C.M.S.
Group, Lasmiditan is an effective acute treatment for migraine: a phase 3
randomized study, Neurology 91 (2018) 2222-2232.
[92] P.J. Goadsby, L.A. Wietecha, E.B. Dennehy, B. Kuca, M.G. Case, S.K. Aurora, C.
Gaul, Phase 3 randomized, placebo-controlled, double-blind study of lasmiditan
for acute treatment of migraine, Brain 142 (2019) 1894-1904.
[93] M.P. Cohen, D.T. Kohlman, S.X. Liang, V. Mancuso, F. Victor, Y.-C. Xu, B.-P.
Ying, D.P. Zacherl, D. Zhang, Preparation of pyridinoylpiperidines as 5-HT1F
agonists, 2003. WO2003084949.
PATENT
https://patents.google.com/patent/WO2020095171A1/en
In one embodiment, the present invention provides a process as depicted in scheme III for the preparation of lasmiditan, a compound of formula I.
R= Cl, OH, OR1
R1 = C1 -C6 alkyl, C1-C6 Alkylaryl
Scheme III
[0044] In one embodiment, the present invention provides a process as depicted in scheme IV for the preparation of lasmiditan, a compound of formula I.
Scheme IV
formula I.
Scheme I
[0079] In one embodiment, the present invention provides a process as depicted in scheme II for the preparation of lasmiditan, a compound of formula I.
Scheme II
formula I.
Scheme V [0092] In one embodiment, the present invention provides a process as depicted in scheme VI for the preparation of lasmiditan, a compound of formula I.
Scheme VI
EXAMPLES
[0162] Example-1: Synthesis of compound of formula V
2,6-Dibromopyridine was dissolved in ammonia and mixture was heated in autoclave at about l20°C to about l50°C till reaction complies. After completion of reaction, the reaction mass was allowed to cool and diluted with water. Product was extracted in dichloromethane which on evaporation gives compound of formula-V in crude stage. The crude compound of formula V was purified in mixture of ethyl acetate and hexane to obtain a pale yellow solid.
[0163] Example-2: Synthesis of compound of formula III
l-Methylisonipecotic acid, Diisopropylethylamine, l-hydroxybenzotriazole and N,O- dimethylhydroxylamine hydrochloride were added in dimethylformamide and the reaction mixture was stirred for about 15 minutes. l-(3-dimethylaminopropyl)-3- ethylcarbodiimide hydrochloride) was then added to the reaction mixture and the reaction mass was stirred at ambient temperature to complete the reaction. The solvent was removed by vacuum distillation. Water was added to obtained residue and the pH of the reaction mixture was adjusted to 8. The obtained aqueous reaction mixture was extracted with dichloromethane and evaporated to get compound of formula III as an oil.
[0164] Example-3: Synthesis of compound of formula II
Methylene chloride was added to 2,4,6-trifluoro benzoic acid, dimethyl formamide was added to the reaction mass and the reaction mass was cooled to about 5°C to l0°C. Oxalyl chloride was added to the reaction mass, the temperature of the reaction mass was raised to 25°C to 30°C and reaction mass was stirred at about 25°C to 30°C for about 2 hours. The solvent was distilled out under vacuum to obtain 2,4,6-trifluoro benzoyl chloride as an oil. Compound of formula V was dissolved in tetrahydrofuran and trimethylamine was added to the obtained reaction mass. The reaction mass was then cooled to about l5°C to 20°C. To the reaction mass was added 2,4,6-trifluoro benzoyl chloride dissolved in tetrahydrofuran. The temperature of the reaction mass was raised to about l5°C to 20°C and the reaction mass was stirred for about 1 hour. The mixture of water and ethyl acetate was added to the reaction mass to obtain organic and aqueous layers. Organic layer was washed with water sodium chloride solution. The solvent was distilled out completely to obtain an oil. The obtained oil was crystallized using diisopropylether. The obtained solid was dried. [0165] Example-4: Synthesis of compound of formula I
The compound of formula II is added to THF and the reaction mass is stirred. N-BuLi is added to the reaction mixture at about -78°C. The mixture is stirred for about 15 to about 30 minutes. The compound of formula-III is added to the reaction mass and the reaction mass is stirred at about -78°C to about -50°C for about 2h. The reaction mass is quenched with aqueous hydrochloric acid and extracted with dichloromethane. The extract is successively washed with water and brine, dried over MgS04 and then concentrated in vacuum to get compound of formula-I.
[0166] Example-5: Synthesis of compound of formula I
l-methylisonipecotic acid hydrochloride is dissolved in methylene chloride and dimethyl formamide is added to the reaction mass. The reaction mass is then cooled to about 5°C to l0°C. Oxalyl chloride is slowly added to the reaction mass and the temperature of the reaction mass is raised to about 25°C to 30°C and the reaction mass is stirred for about 2 hours. The solvent is distilled out to obtain l-methylpiperidine-4-carboxylic acid chloride; the obtain product is then dissolved in tetrahydrofuran and compound of formula II is added to thereaction mass. The reaction mass is stirred and n-butyl lithium is added to the reaction mixture at about -78°C. The mass is stirred for about 15 to 30 minutes. The reaction mass is quenched with aqueous hydrochloric acid and extracted with dichloromethane. The extract is washed with water and brine and dried over MgS04 and then concentrated in vacuum to obtain compound of formula I.
[0167] Example-6: Synthesis of compound of formula X
2,4,6-trifluoro benzoic acid was added to dichloromethane and dimethyl formamide was added to the reaction mass. Oxalyl chloride was added to the reaction mass and the reaction mass was stirred at about 25°C to 30°C for about 30 to 40minutes. The solvent was distilled off to get an oil. The obtained oil was dissolved in dichloromethane to obtain 2,4,6-trifluoro benzoyl chloride. Compound of formula XI was dissolved in dichloromethane and triethylamine was adde to the reaction mass. The reaction mass was then cooled to about 0°C to 5°C. 2,4,6-trifluoro benzoyl chloride in dichloromethane was added to the reaction mass and reaction mass was stirred at about 0°C to 5°C for about 30 minutes. The temperature of the reaction mass was raised to about 25°C 30°C. Aqueous hydrochloric acid was added to the reaction mass and reaction mass was stirred for about l0-l5min and organic and aqueous layer were separated. Organic layer was washed with saturated aqueous sodium bicarbonate and aqueous sodium chloride solution. The organic layer was distilled off to obtain a solid. The obtained solid was crystalized using methanol.
[0168] Example-7: Synthesis of compound of formula IX
Lithium hydroxide was dissolved in water and added to the compound of formula X dissolved in tetrahydrofuran to obtain a reaction mass. The reaction mass was stirred at about 20°C to 25°C for about 20 to 30minutes. Aqueous hydrochloric acid and ethyl acetate were added to the reaction mass and reaction mass was stirred. Aqueous and organic layers were separated. Organic layer was washed with water and aqueous sodium chloride solution. The solvent was distilled off to get solid compound of formula IX.
[0169] Example-8: Synthesis of compound of formula VII
Compound of formula IX was added to dimethyl formamide and triethyl amine and N,O- dimethyl hydroxylamine hydrochloride and HATU were added to the reaction mass the reaction mass was stirred at about 25°C to 30°C for about 5 hours to 6 hours. Water and ethyl acetate were added to the reaction mass and organid and aqueous layers were separated. Aqueous layer was extracted with ethyl acetate. Organic layer washed with water and aqueous sodium chloride solution. The solvent was distilled out completely to obtain an oil; the oil was crystallized using diisopropyl ether to obtain compound of formula VII.
[0170] Example-9: Synthesis of compound of formula I
4-chloro-l -methyl piperidine is added to tetrahydrofuran to obtain a reaction mass. The reaction mass is cooled to about l0°C to 20°C, solution of isopropyl magnesium chloride lithium chloride complex in tetrahydrofuran is added to the reaction mass at about l0°C to 20°C. The reaction mass is stirred for about 30 minutes to 60 minutes. Compound of formula VII is dissolved in tetrahydrofuran and added to the reaction mass. Obtained reaction mass is stirred for about 1 hour to 2 hours. Aqueous ammonium chloride solution and ethyl acetate is added to the reaction mass. The organic and aqueous layers are separated. Organic layer is washed with water and aqueous sodium chloride solution. Organis layer is distilled out to obtain compound of formula I.
[0171] Example-10: Synthesis of compound of formula I
4-chloro-l -methyl piperidine is dissolved in tetrahydrofuran, the reaction mass is cooled to about l0°C to 20°C; isopropyl magnesium chloride lithium chloride complex dissolved in tetrahydrofuran is added to the reaction mass at about lO°C to 20°C. The reaction mass is stirred at about lO°C to 20°C for about 30 minutes to 60 minutes. Compound of formula X dissolved in tetrahydrofuran is added to reaction mass and reaction mass is stirred at about l0°C to 20°C for about 60 minutes to 120 minutes. Aqueous ammonium chloride solution and ethyl acetate is added to the reaction mass, the reaction mass is stirred and aqueous and organic layers are separated. Organic layer is washed with water and aqueous sodium chloride solution. Organic layer is distilled out to obtain compound of formula I.
[0172] Example-11: Synthesis of compound of formula XII
2,4,6-trifluoro benzoic acid was dissolved in dichloromethane and dimethylformamide was added to the reaction mass. The reaction mass was cooled to about 0°C to l0°C, oxalyl chloride was slowly added to the reaction mass and the temperature of the reaction mass was raised to about 20°C to 30°C and stirred for about 30 minutes to 40 minutes. The solvent was distilled off to obtain anoil; the oil was dissolved in tetrahydrofuran to obtained a solution of 2,4,6-trifluoro benzoyl chloride. 2-amino pyridine was dissolved in tetrahydrofuran and triethylamine was added to the reaction mass; a solution of 2,4,6- trifluoro benzoyl chloride in THF was added to the reaction mass and the reaction mass was cooled to about 0°C to 5°C. Water and ethylacetate were addd to the reaction mass and the temperature of reaction mass was raised to about 20°C to 30°C. The reaction mass was stirred and organic and aqueous layers were separated. Organic layer was washed with water and sodium chloride solution. Organic layer was distilled off completely to get an oil, the obtained oil was crystallized using diisopropyl ether and methanol to obtain compound of formula XII.
[0173] Example-12: Synthesis of compound of formula I
Compound of formula XII is dissolved in tetrahydrofuran and N-butyl lithium is added to the reaction mixture at about -78°C. The reation mass is stirred for about 15 minutes to 30 minutes. Compound of formula III is added to the reaction mass and the reaction mass is stirred at about -78°C to -50°C for 2 hours. The reaction mass is quenched with aqueous hydrochloric acid and extracted with dichloromethane. The extract is washed with water and brine, dried over MgS04 and then concentrated in vacuum to get compound of formula I.
[0174] Example-13: Synthesis of compound of formula I l-methylisonipecotic acid hydrochloride is dissolved in methylene chloride and dimethylformamide is added to the reaction mass. The reaction mass is to about 5°C to l0°C and oxalyl chloride is added to the reaction mass, the temperature of the reaction mass is raised to about 25°C to 30°C and reaction mass is stirred for about 2 hours. The solvent is distilled off to obtain l-methylpiperidine-4-carboxylic acid chloride. 1- methylpiperidine-4-carboxylic acid chloride is dissolved in tetrahydrofuran and compound of formula XII dissolved tin tetrahydrofuran is added to it; the reaction mass is stirred. N-butyl lithium is added to the reaction mixture at about -78°C. The mixture is stirred for about 15 minutes to 30 minutes. The reaction mass is quenched with aqueous hydrochloric acid and extracted with dichloromethane. The extract is washed with water and brine, dried over MgS04 and then concentrated in vacuum to get compound of formula I.
[0175] Example-14: Synthesis of compound of formula X
To a mixture of 2,4,6-trifluorobenzoic acid (23. l3g), dichloromethane (260mL) and dimethylformamide (0.2mL) was added oxalyl chloride (25.08g) at about 25°C and the reaction mixture was stirred for about lh. The reaction mixture was concentrated and methylene chloride (75mL) was added to the obtained residue. The resulting solution was added to a mixture of compound of formula XI (15g), methylene dichloride (l50mL) and triethylamine (12.9g) cooled to about 0°C and stirred for about 30min. The temperature of the reaction mixture was raised to about 25°C and 1M aqueous hydrochloric acid was added to it. The two layers were separated and the organic layer was washed with saturated aqueous sodium bicarbonate and aqueous sodium chloride solution. The organic layer was concentrated and the residue was crystallized in methanol and dried. Yield: l8g Mass spectrum: m/z= 309(M-l); ¾ NMR (CDCh) ppm 3.94(s, 3H) 6.69(m, 2H) 7.88(m, 1H) 7.95(t, 1H) 8.6l(d, 1H) 9.34(s, 1H); 13C NMR (CDCh) ppm: 165.10, 164.72, 162.57,
161.80, 161.71, 161.65, 161.56, 159.27, 159.17, 159.12, 159.02, 158.32, 151.06, 145.79,
139.81, 121.76, 118.31, 110.82, 110.77, 101.29, 101.25, 101.03, 100.99, 100.77, 100.74, 52.91
[0176] Example-15: Synthesis of compound of formula X
To a mixture of 2,4,6-trifluorobenzoic acid (20g), methylene chloride (200mL) and dimethylformamide (0.2mL) was added oxalyl chloride (l9.24g) at about 25°C for about lh. The reaction mixture was concentrated and methylene chloride (60mL) was added to the obtained residue. The resulting solution was added to a mixture of compound of formula XI (l2g), methylene chloride (l40mL) and triethylamine (lOg) cooled to about 0°C and stirred for about 30min. The temperature of the reaction mixture was raised to about 25°C and 1M aqueous hydrochloric acid was added it. The two layers were separated and the organic layer was washed with saturated aqueous sodium bicarbonate and aqueous sodium chloride solution. The organic layer was concentrated and the residue was crystalized in methanol and dried. Yield: l8g; HPLC Purity: 99.89%
[0177] Example-16: Synthesis of compound of formula X
To a mixture of 2,4,6-trifluorobenzoic acid (lOg), methylene chloride (lOOmL) and dimethylformamide (0.2mL) was added oxalyl chloride (9.6g) at about 25°C and stirred for about lh. The reaction mixture was concentrated and methylene chloride (50mL) was added to the obtained residue. The resulting solution was added to a compound of formula XI (6g), methylene chloride (50mL) and triethylamine (5g) cooled to about 0°C and stirred for about 30min. The temperature of the reaction mass was raised to about 25°C and 1M aqueous hydrochloric acid was added to it. The two layers were separated and the organic layer was washed with saturated aqueous sodium bicarbonate and aqueous sodium chloride solution. The organic layer was concentrated and the residue was crystallized in methanol and dried. Yield: 9. lg; HPLC Purity: 99.72%
[0178] Example-17: Synthesis of compound of formula X
To a mixture of 2,4,6-trifluorobenzoic acid (5g) in dimethylformamide (25mL) was added compound of formula XI (4.75g) and triethylamine (5.73g) and the reaction mixture was stirred for about l5min. HATU (l3g) was portion wise added to the reaction mixture at about 25°C and the reaction mixture was stirred at about room temperature for about 15h. Water was added to the reaction mixture and the reaction mixture was extracted with ethyl acetate. The organic layer was washed with aqueous hydrochloric acid, aqueous sodium bicarbonate and aqueous sodium chloride solution. The organic layer was concentrated and the residue was crystallized in methanol and dried. Yield: 2.0lg; HPLC Purity: 97.68%
[0179] Example-18: Synthesis of compound of formula X
A mixture of 2,4,6-trifluorobenzoic acid (lOg) and thionyl chloride (30ml) was heated to about 70 to 75°C and maintained for about lh. The reaction mixture was concentrated and methylene chloride (50mL) was added to the obtained residue. The resulting solution was added to a mixture of compound of formula XI (9.5g), triethylamine (l l .47g) and methylene chloride (50ml) cooled to about 0°C and stirred for about lh. The temperature of the reaction mass was raised to about 25°C and 1M aqueous hydrochloric acid (lOOmL) was added to the reaction mixture. The two layers were separated and the organic layer was washed with saturated aqueous sodium bicarbonate and aqueous sodium chloride solution. The organic layer was concentrated and the residue was crystallized in methanol and dried. Yield: 8.7g; HPLC Purity: 92.64%
[0180] Example-19: Synthesis of compound of formula IX
To a solution of lithium hydroxide (3.35g) in water (75mL), was added compound of formula X (15g) and tetrahydrofuran (150mL) at about 25°C and the reaction mixture was stirred for about 15h. Aqueous hydrochloric acid and ethyl acetate were added to the reaction mixture and the two layers were separated. The organic layer was washed with aqueous sodium chloride solution. The organic layer was concentrated and the residue was crystallized in diisopropyl ether. Yield: 12.7g
Mass spectrum: m/z= 297(M+l); ¾ NMR (DMSO) ppm 7.33(m, 2H), 7.86(d, 1H), 8.05(t, 1H), 8.39(d, 1H), l l .74(s, 1H), l3.33(s, 1H)
13C NMR (CDCb) ppm: 166.19, 164.50, 162.02, 161.26, 161.10, 160.99, 159.27, 158.78, 158.67, 158.62, 158.51, 151.61, 147.65, 140.37, 121.69, 117.81, 112.42, 101.79, 101.52, 101.49, 101.22
[0181] Example-20: Synthesis of compound of formula IX
To a solution of lithium hydroxide (2.35g) in water (75mL), was added compound of formula X (15g) and tetrahydrofuran (150mL) at about 25°C and the reaction mixture was stirred for about 15h. Aqueous hydrochloric acid and ethyl acetate were added to the reaction mixture and the two layers were separated. The organic layer was washed with aqueous sodium chloride solution. The organic layer was concentrated and the residue was crystallized in diisopropyl ether. Yield: 13. lg; HPLC Purity: 97.64%
[0182] Example-21: Synthesis of compound of formula IX
To a solution of sodium hydroxide (7.74g) in water (l50mL), was added compound of formula X (30g) and tetrahydrofuran (300mL) at about 25°C and the reaction mixture was stirred for about 2h. Aqueous hydrochloric acid and ethyl acetate were added to the reaction mixture and the two layers were separated. The organic layer was washed with aqueous sodium chloride solution. The organic layer was concentrated and the residue was crystallized in diisopropyl ether. Yield: 27.3g; HPLC Purity: 98.14%
[0183] Example-22: Synthesis of compound of formula VII
To a mixture of compound of formula IX (lOg) in dimethylformamide (40mL) was added and triethylamine (l0.23g), Af/7-dimethyl hydroxyl amine hydrochloride (3.22g) and HATU (l5.4g) and the reaction mixture was stirred at about 25 °C for about 15h. Water, ethyl acetate and tetrahydrofuran were added to the reaction mixture and the two layers were separated. The aqueous layer was extracted with ethyl acetate and tetrahydrofuran. The combined organic layer was washed with aqueous sodium bicarbonate, water and aqueous sodium chloride solution. The organic layer was concentrated and the residue was crystallized in ethyl acetate. Yield: 5.5g
Mass spectrum: m/z= 340(M+l); 1HNMR (CDCb) ppm 3.37(s, 3H), 3.69(s, 3H), 6.80(m, 2H), 7.44(d, 1H), 7.87(t, 1H), 8.42(d, 1H), 8.53(s, 1H)
13C NMR (CDCb) ppm: 165.41, 165.26, 165.10, 162.87, 162.72, 162.57, 162.13, 162.04, 161.98, 61.89, 159060, 159.50, 159.45, 159.35, 157.97, 151.18, 149.79, 139.31, 119.74, 115.61, 110.73, 110.54, 110.49, 110.35, 101.62, 101.58, 101.36, 101.32, 101.10, 101.06, 61.50
[0184] Example-23: Synthesis of compound of formula VII
To a mixture of compound of formula IX (20g) in dimethylformamide (l25mL), was added triethylamine (25.6g), Af/7-dimethyl hydroxyl amine hydrochloride (9.05g) and HATU (38.5g) and the reaction mixture was stirred at about 25 °C for about 15h. Water, ethyl acetate and tetrahydrofuran were added to the reaction mixture and the two layers were separated. The aqueous layer was extracted with ethyl acetate and tetrahydrofuran. The combined organic layer was washed with aqueous sodium bicarbonate and aqueous sodium chloride solution. The organic layer was concentrated and the residue was crystallized in ethyl acetate. Yield: 20.2g; HPLC Purity: 99.35%
[0185] Example-24: Synthesis of compound of formula VII
To a mixture of compound of formula IX (lOg) in dimethylformamide (50mL), was added triethylamine (l0.23g), Af,U-dimethyl hydroxylamine hydrochloride (3.6g) and HATU (l5.4g) and the reaction mass was stirred at about 25°C for about 3h. Water was added to the reaction mixture. The reaction mixture was stirred at about 25°C for about 30min and filtered. The solid was washed with water and dried under vacuum. Yield: l0.07g; HPLC Purity: 99.33%
[0186] Example-25: Synthesis of lasmiditan hemisuccinate
(a) To a mixture of 4-chloro-l-methylpiperidine hydrochloride (25g) in water (75mL) was added potassium carbonate (25g). The reaction mixture was extracted with methyl
/cvv-butyl ether. The organic layer was washed with aqueous sodium chloride solution and concentrated to give 4-chloro-l-methylpiperidine free base. Yield: l4g
(b) To a mixture of magnesium turnings (12.5g), pinch of iodine and ethylene dibromide (lmL) in tetrahydrofuran (40mL) was added a mixture of 4-chloro-l -methyl piperidine (20g) in tetrahydrofuran (lOOmL). The reaction mixture was heated to reflux for about 2h and cooled to about 25°C. The reaction mixture was added to a mixture of compound of formula VII (lOg) in tetrahydrofuran (l50mL) at about 0°C. The reaction mixture was stirred for about lh at about 0°C and aqueous ammonium chloride solution and MTBE was added to it. The temperature of the reaction mixture was raised to about 25°C and the two layers were separated. The organic layer was washed with aqueous sodium chloride solution and concentrated. The residue was dissolved in ethanol (lOOmL) and succinic acid (3.5g) was added to it at about 25°C. The reaction mixture was heated to about 75°C. The reaction mixture was cooled to about 20°C and stirred overnight. The solid obtained was filtered, washed with ethanol and dried under vacuum. Yield: 6.5g
[0187] Example-26: Synthesis of lasmiditan hemisuccinate
To a mixture of compound of formula VII (5g) in tetrahydrofuran (50mL) cooled to about 0°C, was slowly added 1M solution of l-methylpiperidine-4-yl-magnesium chloride in THF (75mL) and the reaction mixture was stirred for about lh. Water was added slowly at about 0-5°C and then ethyl acetate and the temperature was raised to about 25°C. The two layers were separated and the organic layer was washed with aqueous sodium chloride solution and concentrated. The residue was dissolved in isopropyl alcohol (50mL) and succinic acid (1 75g) was added to it at about 25°C. The reaction mixture was heated to about 75°C. The reaction mixture was cooled to about 20°C and stirred for 2h. The solid obtained was filtered, washed with isopropyl alcohol and dried. Yield: 4. l2g; HPLC purity: 99.10%
[0188] Example-27: Synthesis of lasmiditan hemisuccinate To a mixture of compound of formula VII (lOg) in tetrahydrofuran (50mL) cooled to about 0°C, was added 1M solution of l-methylpiperidine-4-yl-magnesium chloride in tetrahydrofuran (l80mL) slowly at about 0°C and the reaction mixture was stirred for about 2h. 10% aqueous ammonium chloride solution and ethyl acetate were added to the reaction mixture at about 0-5°C and the temperature was raised to about 25°C. The two layers were separated. The organic layer was washed with aqueous sodium chloride solution and concentrated. The residue was dissolved in isopropyl alcohol (lOOmL) and succinic acid (3.4g) was added to it at about 25°C. The reaction mixture was heated to about 70°C. The reaction mixture was cooled to about 20°C and stirred for 30min. The solid obtained was filtered, washed with isopropyl alcohol and dried. Yield: 9g; HPLC Purity: 99.45%
[0189] Example-28: Synthesis of lasmiditan hemisuccinate
To a mixture of compound of formula VII (8g) in tetrahydrofuran (50mL) cooled to about 0°C, was added 1M solution of l-methylpiperidine-4-yl-magnesium chloride in tetrahydrofuran (H8mL) slowly at about 0°C and the reaction mixture was stirred for about lh. 10% aqueous hydrochloric acid and ethyl acetate were added to the reaction mixtures at about 0-5°C and the temperature was raised to about 25°C. The two layers were separated and the organic layer was washed with aqueous sodium chloride solution and concentrated. The residue was dissolved in isopropyl alcohol (80ml) and succinic acid (2.73g) was added to it at about 25°C. The reaction mixture was heated to about 70°C. The reaction mixture was cooled to about 20°C and stirred for about lh. The solid obtained was filtered, washed with isopropyl alcohol and dried. Yield: 6.35g; HPLC Purity: 98.25%
[0190] Example-29: Synthesis of lasmiditan hemisuccinate
To a mixture of compound of formula VII (l8g) in tetrahydrofuran (l80mL) cooled to about 0°C to about -l0°C, was added 1M solution of l-methylpiperidine-4-yl-magnesium chloride in tetrahydrofuran (l80mL) slowly at about -5°C to about -l0°C and the reaction mixture was stirred for about lh. Water was added slowly at about 0-5°C and then ethyl acetate and the temperature was raised to about 25°C. The two layers were separated and the organic layer was washed with aqueous sodium chloride solution and concentrated. The residue was dissolved in isopropyl alcohol (l80mL) and succinic acid (6g) was added to it at about 25°C. The reaction mixture was heated to about 50°C. The reaction mixture was cooled to about 20°C. The solid was filtered, washed with isopropyl alcohol and dried. Yield: 14.6g
[0191] Example-30:
A mixture of lasmiditan hemisuccinate (3g) in isopropyl alcohol (30mL) was heated to about 65-70°C and stirred for about 30min. The reaction mixture was cooled to about 25°C and stirred for about lh. The solid obtained was filtered, washed with isopropyl alcohol and dried. Yield: 2.6g; HPLC Purity: 99.38%
XRPD peaks of lasmiditan hemisuccinate
To a mixture of lasmiditan hemisuccinate (8g) in water (80mL) was added 10% aqueous sodium carbonate solution (40mL) at about 25°C followed by addition of ethyl acetate (80mL). The two layers were separated and the organic layer was washed with aqueous sodium chloride solution and concentrated. The residue was dissolved in isopropyl alcohol (40mL) at about 45°C and succinic acid (2.59g) in isopropyl alcohol (40mL) was added to it. The reaction mixture was cooled to about 25°C and stirred for about lh. The solid was filtered, washed with isopropyl alcohol and dried. Yield: 5.45g; HPLC Purity: 99.69% [0193] Example-32:
A mixture of lasmiditan hemisuccinate (5g) in isopropyl alcohol (50mL) was heated to about 65-70°C and stirred for about 30min. The mixture was cooled to about 25°C and stirred for about lh. The solid was filtered, washed with isopropyl alcohol and dried. Yield: 4.7g; HPLC purity: 99.77%
The following examples follow similar process as described in Example-32.
Mechanism of action
Lasmiditan is a serotonin receptor agonist that, like the unsuccessful LY-334,370, selectively binds to the 5-HT1F receptor subtype. A number of triptans have been shown to act on this subtype as well, but only after their affinity for 5-HT1B and 5-HT1D has been made responsible for their anti-migraine activity.[7] The lack of affinity for these receptors might result in fewer side effects related to vasoconstriction compared to triptans in susceptible people, such as those with ischemic heart disease, Raynaud’s phenomenon or after a myocardial infarction,[8] although a 1998 review has found such side-effects to rarely occur in people taking triptans.[9][10]
Adverse effects
There is a risk of driving impairment while taking lasmiditan. People are advised not to drive or operate machinery for at least eight hours after taking lasmiditan, even if they feel well enough to do so. People who cannot follow this advice are advised not to take lasmiditan. The drug causes central nervous system (CNS) depression, including dizziness and sedation. It should be used with caution if taken in combination with alcohol or other CNS depressants.[2]
History
Lasmiditan was discovered by Eli Lilly and Company and was then relicensed to CoLucid Pharmaceuticals in 2006, until CoLucid was bought by Eli Lilly in 2017, to allow Eli Lilly to reacquire the drug’s intellectual property.[11] The drug is protected by patents until 2031.[12]
Phase II clinical trials for dose finding purposes were completed in 2007, for an intravenous form[13] and in early 2010, for an oral form.[14] Eli Lilly submitted a new drug application to the U.S. Food and Drug Administration (FDA) in November 2018.[15]
Three Phase III clinical trials were completed. The SPARTAN trial compared placebo with 50, 100, and 200 mg of lasmiditan.[16] SAMURAI compared placebo with 100 and 200 mg doses of lasmiditan. GLADIATOR is an open-label study that compared 100 and 200 mg doses of lasmiditan in subjects that received the drug as part of a prior trial.[17]
Topline results from the SPARTAN trial showed that the drug induced met its primary and secondary endpoints in the trial. The primary result showed a statistically significant improvement in pain relief relative to placebo 2 hours after the first dose. The secondary result showed a statistically significantly greater percentage of subjects were free of their most bothersome symptom (MBS) compared with placebo at two hours following the first dose.[18]
The FDA approved lasmiditan primarily based on data from two clinical trials, Trial 1 (# NCT02439320) and Trial 2 (#NCT02605174) of 4439 subjects with migraine headaches with or without aura.[19] Trials were conducted at 224 sites in the United States, the United Kingdom, and Germany.[19]
The FDA approved the drug in October 2019.[19] However, as of October 2019, the drug was awaiting Drug Enforcement Administration (DEA) scheduling before it was made available in the United States.[20] It was placed into Schedule V in January 2020.[21][1]
Dosage
Lasmiditan is delivered in 50 & 100 mg tablet form.[22]
Novel crystalline forms of a 5-HT1F receptor agonist, particularly lasmiditan – designated as Forms 1-3 and A-D – processes for their preparation and compositions comprising them are claimed. Also claim is their use for treating anxiety, fatigue, depression, premenstrual syndrome, trauma syndrome, memory loss, dementia (including Alzheimer’s), autism, schizophrenia, attention deficit hyperactivity disorder, obsessive-compulsive disorder, epilepsy, anorexia nervosa, alcoholism, tobacco abuse, mutism and trichotillomania.
Biological Activity
Lasmiditan (also known as COL-144 and LY573144) is a high-affinity, highly selective serotonin (5-HT) 5-HT(1F) receptor agonist.
In vitro binding studies show a K(i) value of 2.21 nM at the 5-HT(1F) receptor, compared with K(i) values of 1043 nM and 1357 nM at the 5-HT(1B) and 5-HT(1D) receptors, respectively, a selectivity ratio greater than 470-fold. Lasmiditan showed higher selectivity for the 5-HT(1F) receptor relative to other 5-HT(1) receptor subtypes than the first generation 5-HT(1F) receptor agonist LY334370.
In two rodent models of migraine, oral administration of lasmiditan potently inhibited markers associated with electrical stimulation of the trigeminal ganglion (dural plasma protein extravasation, and induction of the immediate early gene c-Fos in the trigeminal nucleus caudalis).
Conversion of different model animals based on BSA (Value based on data from FDA Draft Guidelines)
| Species |
Mouse |
Rat |
Rabbit |
Guinea pig |
Hamster |
Dog |
| Weight (kg) |
0.02 |
0.15 |
1.8 |
0.4 |
0.08 |
10 |
| Body Surface Area (m2) |
0.007 |
0.025 |
0.15 |
0.05 |
0.02 |
0.5 |
| Km factor |
3 |
6 |
12 |
8 |
5 |
20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by |
Animal B Km |
| Animal A Km |
For example, to modify the dose of resveratrol used for a mouse (22.4 mg/kg) to a dose based on the BSA for a rat, multiply 22.4 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for resveratrol of 11.2 mg/kg.


PATENT
WO 03084949
https://www.google.co.in/patents/WO2003084949A1?cl=en
8. 2,4,6-Trifluoro-N-[6-(l -methyl-piperidin-4-ylcarbonyl)-pyridin-2-yl]- benzamide mono-hydrochloride salt
Combine 2-amino-6-(l-methylpiperidin-4-ylcarbonyl)pyridine (0.20 g, 0.92 mmol), 2,4,6-Trifluorobenzoyl chloride (0.357 g, 1.84 mmol), and 1 ,4-Dioxane (10 mL), and stir while heating at reflux. After 3 hr., cool the reaction mixture to ambient temperature and concentrate. Load the concentrated mixture onto an SCX column (lOg), wash with methanol, and elute with 2M ammonia in methanol. Concentrate the eluent to obtain the free base of the title compound as an oil (0.365 g (>100%)). Dissolve the oil in methanol (5 mL) and treat with ammonium chloride (0.05 g, 0.92 mmol). Concentrate the mixture and dry under vacuum to obtain the title compound. HRMS Obs. m/z 378.1435, Calc. m/z 378.1429; m.p. 255°C (dec).
Examples
21. 2,4,6-Trifluoro-N-[6-(l-methyl-piperidin-4-ylcarbonyl)-pyridin-2-yl]- benzamide

Add triethylamine (10.67 mL, 76.70 mmol, 2.4 eq) to a solution of 2-amino-(6-(l- methylpiperidin-4-ylcarbonyl)-pyridine (7g, 31.96 mmol, 1 eq) in anhydrous THF (100 mL) under a nitrogen atmosphere. Add 2,4,6-triflubenzoylchloride (7.46g, 5 mL, 38.35 mmol, 1.20 eq) dropwise at room temperature. After 2 hrs., add additional 2,4,6- triflubenzoylchloride (0.75 mL, 0.15 eq) and triethylamine (1.32 mL, 0.3 eq) to the reaction mixture and agitate the mixture for an additional 3 hrs. Quench the reaction with distilled water (10 mL) and 30%o NaOH (15 mL). Stir the resulting biphasic system for 1 hour and then separate the phases. Extract the organic fraction by adding H2O (75 mL) and acetic acid (12 mL), followed by cyclohexane (70 mL). Wash the organic fraction with H2O (50 mL) containing acetic acid (1 mL). Combine all the aqueous fractions and washes and neutralize the mixture with 30% NaOH (15 mL). Extract with methyl-tert- butyl ether (MTBE) (3×50 mL). Combine the organic fractions and dry with MgSO4, filter, concentrate under reduce pressure, and vacuum dry at room temperature, to obtain the title compound as a light-brown solid (11.031 g, 91 % yield).
Mass spectrum, (Electrospray) m/z = 378 (M+l); Η NMR (250 MHz, Chloroform-D) ppm 1.54 (m, 2 H) 2.02 (m, 2 H) 2.13 (t, J=l 1.48 Hz, 2 H) 2.29 (s, 3 H) 2.80 (m, J=l 1.96 Hz, 1 H) 3.56 (m, 1 H) 4.26 (d, J=7.87 Hz, 1 H) 6.17 (d, J=8.50 Hz, 1 H) 6.75 (m, 2 H) 7.45 (t, J=7.87 Hz, 1 H) 7.53 (m, 1 H) 7.95 (s, 1 H); 13C-NMR: (62.90 MHz, Chloroform-D) ppm 202.78; 162.6 (dm C-F-couplings); 162.0 (m C-F-couplings); 160.1 (m C-F-couplings); 158.1 ; 150.0; 139.7; 1 19.3; 1 17.9; 1 10.2 (m C-F-couplings); 100.9 (m C-F-couplings); 55.2; 46.5; 41.9; 28.1
22. 2,4,6-Trifluoro-N-[6-(l-methyl-piperidin-4-ylcarbonyl)-pyridin-2-yl]- benzamide mono-hydrochloride salt

Dissolve 2,4,6-trifluoro-N-[6-(l-methylpiperidin-4-ylcarbonyl)-pyridin-2-yl]- benzamide – free base (5g, 23.26mmol) in isopropanol (50 mL) at room temperature and add a solution of 3.3 M diethylether/HCl (8 mL). Heat the reaction mixture under reflux for 30 minutes. Cool the reaction mixture to room temperature and agitate for 2 hrs. Filter the resulting white precipitate and rinse with isopropanol (5 mL). Dry the residual solid under reduce pressure at 40°C overnight to obtain the title compound (5.12 g, 93% yield). M.p. 223-224°C (sublimation); Η NMR (400 MHz, d6-DMSO) d ppm 1.94 (m, 2 H) 2.14 (m, J=11.15 Hz, 2 H) 2.74 (s, 3 H) 2.99 (m, J=9.19 Hz, 2 H) 3.49 (m, J=1 1.15 Hz, 2 H) 3.77 (m, 1 H) 7.41 (t, J=8.71 Hz, 2 H) 7.78 (d, J=7.43 Hz, 1 H) 8.10 (t, J=7.92 Hz, 1 H) 8.37 (d, J=6.85 Hz, 1 H) 10.50 (s, 1 H) 1 1.51 (s, 1 H); 13C-NMR: (100.61 MHz, Chloroform-D) ppm 200.7; 130.6-158.0 (m, C-F-couplings); 150.4; 150.1; 140.2; 118.5; 1 18.2; 11 1.9; 101.3 (t, C-F couplings); 52.8; 42.6; 25.2
23. 2,4,6-Trifluoro-N-[6-(l-methyl-piperidine-4-carbonyl)-pyridin-2-yl]- benzamide hemi-succinate salt
Add succinic acid (0.25g, 2.148 mmol, 0.5eq) to a solution of 2,4,6-trifluoro-N-[6-
(l-methyl-piperidin-4-ylcarbonyl)-pyridin-2-yl]-benzamide – free base (1.62g, 4.297 mmol, leq) in acetone (16.2 mL), at room temperature. Warm the solution under reflux for 30 minutes. Cool the solution to room temperature and filter off the resulting white precipitate. Rinse the precipitate with acetone (0.2 mL) and dry under vacuum at 50°C for 16 hours to provide the title compound (1.5g, 80% yield). M.p. 198.5°C; mass spectrum (Electrospray) m/z = 495.45
The following examples are prepared by combinatorial chemistry techniques as follows:
Examples 24-54
Combine R-acid (300 μL of 0.5M solution in dimethylformamide (DMF)), HATU (57 mg, 0.15 mmol), collidine (19 μL, 0.15 mmol), 2-amino-(6-(l-methylpiperidin-4- ylcarbonyl)-pyridine and DMF (1.5 mL), and agitate for 48 hr. Dilute the reaction mixture with 10% acetic acid in methanol (0.5 L). Load the resulting reaction mixture onto a 2 g SCX column. Wash the column thoroughly with methanol and then elute with 1 M ammonia in methanol. Concentrate the eluent and further purify the product by high- throughput mass guided chromatography. This procedure is repeated in parallel for examples 24-54.
Examples 55-58
Heat R-acid chloride (300 μL of 0.5M solution in pyridine) to 55°C, add 2-amino- (6-(l-methylpiperidin-4-ylcarbonyl)-pyridine (200 μL of 0.5M solution in pyridine), and continue heating the reaction mixture for 24 hr. Concentrate the reaction mixture and then dilute with 10% Acetic acid in methanol (0.5 mL) and methanol (0.5 mL). Load the resulting reaction mixture directly onto a 2 g SCX column. Thoroughly wash the column with methanol and then elute the column with 1 M ammonia in methanol. Concentrate the eluent and then further purify the product by high- throughput mass guided chromatography. This procedure is repeated in parallel for examples 55-58.
Examples 59-71
Heat 2-amino-(6-(l-methylpiperidin-4-ylcarbonyl)-pyridine (200 μL of 0.5M solution in pyridine) to 55°C then add R-acid chloride (0.10 mmol), heat for 2 hr. Concentrate the reaction mixture and then dilute with 10% Acetic acid in methanol (0.5 mL) and methanol (0.5 mL). Load the resulting reaction mixture directly onto a 2 g SCX column. Thoroughly wash the column with methanol and then elute the column with 1 M ammonia in methanol. Concentrate the eluent and then further purify the product by high-throughput mass guided chromatography. This procedure is repeated in parallel for examples 59-71.
PATENT
WO 2018010345
Lasmiditan, also known as COL-144, LY573144, is a 5-HT 1F receptor agonist. Can be used to inhibit neuronal protein extravasation, to treat or prevent migraine in patients with diseases or conditions associated with other 5-HT 1F receptor dysfunction. The chemical name is 2,4,6-trifluoro-N- [6 – [(1 -methylpiperidin-4-yl) carbonyl] -pyridin- 2-yl] -benzamide, which has the chemical structure shown below I) shows:
Lasmiditan is a new and selective 5-HT 1F receptor agonist. It acts against migraine and other 5-HT 1F receptor related diseases by enhancing 5-HT 1F receptor activation while avoiding vasoconstrictive activity and inhibiting neuronal protein extravasation such as Migraine (including migraine, migraine headache, neurovascular headache), general pain, trigeminal neuralgia, anxiety, panic disorder, depression, post traumatic syndrome, dementia and the like.
Patent document CN100352817C reports on Lasmiditan, Lasmiditan hemisuccinate and Lasmiditan hydrochloride and the synthetic preparation thereof, and discloses the mass spectra of Lasmiditan, Lasmiditan hemisuccinate and Lasmiditan hydrochloride, 1 H-NMR, 13 C -NMR detection data and the melting points of Lasmiditan hemisuccinate and Lasmiditan hydrochloride. The inventor of the present invention has found that Lasmiditan, which is obtained according to the preparation method of Example 17 and Example 21 in CN100352817C, is a light brown oily amorphous substance, which has the defects of instability, moisture absorption and poor morphology.
Example 8 of patent document CN100352817C reports the preparation of Lasmiditan hydrochloride, which mentions Lasmiditan free base as an oily substance. The Lasmiditan hydrochloride obtained according to the preparation method of Example 8 in CN100352817 is a white amorphous substance which also has the disadvantages of unstable crystalline form, high hygroscopicity and poor topography.
The synthesis of Lasmiditan hemisuccinate intermediate, including Lasmiditan and Lasmiditan hydrochloride, is reported in Example 2 of U.S. Patent No. 8,697,876 B2. The inventor’s study found that Lasmiditan prepared according to US8697876B2 is also a pale brown oily amorphous substance and Lasmiditan hydrochloride is also a white amorphous substance.
In view of the deficiencies in the prior art, there is still a need in the art for the development of crystalline polymorphic Lasmiditan solid forms with more improved properties to meet the rigorous requirements of pharmaceutical formulations for physico-chemical properties such as morphology, stability and the like of active materials.
Preparation 1 Preparation of Lasmiditan (Prior Art)
Lasmiditan was prepared as described in Example 21 of CN100352817C by the following procedure: Triethylamine (10.67 mL, 76.70 mmol, 2.4 equiv) was added to a solution of 2-amino- (6- (1-methylpiperidine -4-yl) -carbonyl) -pyridine (7 g, 31.96 mmol, 1 eq) in dry THF (100 mL). 2,4,6-Trifluorobenzoyl chloride (7.46 g, 5 mL, 38.35 mmol, 1.20 equiv.) Was added dropwise at room temperature. After 2 hours, an additional 2,4,6-trifluorobenzoyl chloride (0.75 mL, 0.15 eq) and triethylamine (1.32 mL, 0.3 eq) were added to the reaction mixture and the mixture was stirred for a further 3 h. The reaction was quenched with distilled water (10 mL) and 30% NaOH (15 mL). The resulting two-phase system was stirred for 1 hour, then the two phases were separated. By addition of H 2 to extract the organic portion O (75mL) and acetic acid (12mL), followed by addition of cyclohexane (70mL). The organic portion was washed with water (50 mL) containing acetic acid (1 mL). All aqueous phases were combined, washed and neutralized with 30% NaOH (15 mL). Extract with methyl tert-butyl ether (MTBE) (3 x 50 mL). The organic phases were combined, dried MgS04 . 4 dried, filtered, and concentrated under reduced pressure and dried in vacuo at room temperature to give the title compound as a pale brown solid (11.031g, 91% yield).
The 1 H-NMR (CDCl 3 ) data of the product are as follows:
1 H NMR (400 MHz, CHLOROFORM-D) ppm 1.54 (m, 2H) 2.02 (m, 2H) 2.13 (t, J = 18.37 Hz, 2H) 2.29 (s, 3.56 (d, J = 12.59 Hz, 1H) 6.17 (d, J = 13.6 Hz, 1H) 6.75 (m, 2H) 7.45 (t, J = 12.59 Hz, 1H) 7.53 (m, 1H ) 7.95 (s, 1H).
The isothermal adsorption curve shown in Figure 5, in the 0% to 80% relative humidity range of 9.5% weight change.
The above characterization results show that Lasmiditan obtained by the preparation method of Example 21 according to CN100352817C is amorphous.
Preparation 2 Preparation of Lasmiditan hydrochloride (Prior Art)
The Lasmiditan hydrochloride was prepared as described in Example 8 of CN100352817C by the following procedure: A mixture of 2-amino-6- (1-methylpiperidin-4-yloxy) pyridine Trifluorobenzoyl chloride (3.57 g, 18.4 mmol) and 1,4-dioxane (100 mL) were combined and heated to reflux with heating. After 3 hours, cool the reaction mixture to room temperature, reduce pressure and concentrate. The concentrated mixture was loaded onto a SCX column (10 g), washed with methanol and eluted with 2M ammonia in methanol. The eluate was concentrated to give the title compound as an oily free base (3.65 g (> 100%)). The oil was dissolved in methanol (50 mL) and treated with ammonium chloride (0.5 g, 9.2 mmol). The mixture was concentrated and dried in vacuo to give a white amorphous.
IC characterization showed that Lasmiditan hydrochloride salt formed by Lasmiditan and hydrochloric acid in a molar ratio of 1: 1.
The XRPD pattern shown in Figure 19, no diffraction peaks, no amorphous.
The PLM pattern is shown in Figure 20 as an irregular, unpolarized solid.
The isotherm adsorption curve is shown in FIG. 21, with a weight change of 8.1% in a relative humidity range of 0% to 80%.
The above characterization results show that: Lasmiditan hydrochloride obtained by the preparation method of Example 8 with reference to CN100352817C is amorphous.
Take 500mg of Lasmiditan of Preparation 1, add 1mL methanol solution containing 5% water to clarify, evaporate the crystals at room temperature and evaporate dry after 1 day to obtain 487mg Lasmiditan Form 1 in 95% yield.
References
- ^ Jump up to:a b “2020 – Placement of Lasmiditan in Schedule V”. DEA Diversion Control Division. 31 January 2020. Retrieved 31 January 2020.
- ^ Jump up to:a b c d “Reyvow- lasmiditan tablet”. DailyMed. 11 October 2019. Retrieved 15 November 2019.
- ^ Jump up to:a b c “FDA approves new treatment for patients with migraine”. U.S. Food and Drug Administration (FDA) (Press release). 11 October 2019. Archived from the original on 16 November 2019. Retrieved 17 October 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
- ^ “Lasmiditan (Professional Patient Advice)”. Drugs.com. 4 June 2019. Retrieved 23 February 2020.
- ^ “Lilly’s Reyvow (lasmiditan) C-V, the First and Only Medicine in a New Class of Acute Treatment for Migraine (ditan), Now Available for Prescription”. Eli Lilly and Company. 31 January 2020. Retrieved 23 February 2020.
- ^ “New Drug Therapy Approvals 2019”. U.S. Food and Drug Administration. 31 December 2019. Retrieved 15 September 2020.
- ^ Rissardo, JamirPitton; Fornari Caprara, AnaLetícia (2020). “The ditans, a new class for acute migraine: Minireview”. Journal of Current Research in Scientific Medicine. 6 (1): 11. doi:10.4103/jcrsm.jcrsm_45_19. ISSN 2455-3069.
- ^ “Molecule of the Month July 2010: Lasmiditan hydrochloride”. Prous Science. Archived from the original on 28 July 2011. Retrieved 3 August 2011.
- ^ Dahlöf CG, Mathew N (October 1998). “Cardiovascular safety of 5HT1B/1D agonists–is there a cause for concern?”. Cephalalgia. 18 (8): 539–45. doi:10.1046/j.1468-2982.1998.1808539.x. PMID 9827245. S2CID 30125923.
- ^ Mutschler E, Geisslinger G, Kroemer HK, Schäfer-Korting M (2001). Arzneimittelwirkungen (in German) (8th ed.). Stuttgart: Wissenschaftliche Verlagsgesellschaft. p. 265. ISBN 978-3-8047-1763-3. OCLC 47700647.
- ^ “Lilly buys migraine biotech CoLucid, and the drug it outlicensed, for $960M”.
- ^ “Lasmiditan – Eli Lilly and Company – AdisInsight”.
- ^ “A Placebo-Controlled Adaptive Treatment Assignment Study of Intravenous COL-144 in the Acute Treatment of Migraine”. ClinicalTrials.gov. 8 November 2019. Retrieved 23 February 2020.
- ^ “Dose-ranging Study of Oral COL-144 in Acute Migraine Treatment”. ClinicalTrials.gov. 20 December 2019. Retrieved 23 February 2020.
- ^ “Lilly Submits New Drug Application to the FDA for Lasmiditan for Acute Treatment of Migraine, Receives Breakthrough Therapy Designation for Emgality (galcanezumab-gnlm) for Prevention of Episodic Cluster Headache”. Eli Lilly and Company. 14 November 2018. Retrieved 12 October 2019 – via PR Newswire.
- ^ Clinical trial number NCT02605174 for “Three Doses of Lasmiditan (50 mg, 100 mg and 200 mg) Compared to Placebo in the Acute Treatment of Migraine (SPARTAN)” at ClinicalTrials.gov
- ^ Clinical trial number NCT02565186 for “An Open-label, Long-term, Safety Study of Lasmiditan for the Acute Treatment of Migraine (GLADIATOR)” at ClinicalTrials.gov
- ^ “Lilly Announces Positive Results for Second Phase 3 Study of Lasmiditan for the Acute Treatment of Migraine”. Archived from the original on 5 August 2017. Retrieved 5 August 2017.
- ^ Jump up to:a b c “Drug Trials Snapshots: Reyvow”. U.S. Food and Drug Administration (FDA). 11 October 2019. Retrieved 26 January 2020.
- ^ Vinluan F (11 October 2019). “FDA OKs Lilly’s Lasmiditan, First New Acute Migraine Drug in Decades”. Xconomy. Retrieved 12 October 2019.
- ^ “Schedules of Controlled Substances: Placement of Lasmiditan in Schedule V”. Federal Register. 31 January 2020.
- ^ “Reyvow (Lasmiditan Tablets): Uses, Dosage, Side Effects, Interactions, Warning”. RxList. Retrieved 20 August 2020.
-
External links
- “Lasmiditan”. Drug Information Portal. U.S. National Library of Medicine.
- /////////////LASMIDITAN, phase III, LILY, COL-144 , LY-573144, CoLucid Pharmaceuticals, PHASE 3, MIGRAINE, ласмидитан, لاسميديتان , FDA 2019
- Capi M, de Andres F, Lionetto L, Gentile G, Cipolla F, Negro A, Borro M, Martelletti P, Curto M: Lasmiditan for the treatment of migraine. Expert Opin Investig Drugs. 2017 Feb;26(2):227-234. doi: 10.1080/13543784.2017.1280457. [Article]
- Nelson DL, Phebus LA, Johnson KW, Wainscott DB, Cohen ML, Calligaro DO, Xu YC: Preclinical pharmacological profile of the selective 5-HT1F receptor agonist lasmiditan. Cephalalgia. 2010 Oct;30(10):1159-69. doi: 10.1177/0333102410370873. Epub 2010 Jun 15. [Article]
- Lupi C, Benemei S, Guerzoni S, Pellesi L, Negro A: Pharmacokinetics and pharmacodynamics of new acute treatments for migraine. Expert Opin Drug Metab Toxicol. 2019 Mar;15(3):189-198. doi: 10.1080/17425255.2019.1578749. Epub 2019 Feb 12. [Article]
- Vila-Pueyo M: Targeted 5-HT1F Therapies for Migraine. Neurotherapeutics. 2018 Apr;15(2):291-303. doi: 10.1007/s13311-018-0615-6. [Article]
- Rubio-Beltran E, Labastida-Ramirez A, Haanes KA, van den Bogaerdt A, Bogers AJJC, Zanelli E, Meeus L, Danser AHJ, Gralinski MR, Senese PB, Johnson KW, Kovalchin J, Villalon CM, MaassenVanDenBrink A: Characterization of binding, functional activity and contractile responses of the selective 5-HT1F receptor agonist lasmiditan. Br J Pharmacol. 2019 Aug 16. doi: 10.1111/bph.14832. [Article]
- Reuter U, Israel H, Neeb L: The pharmacological profile and clinical prospects of the oral 5-HT1F receptor agonist lasmiditan in the acute treatment of migraine. Ther Adv Neurol Disord. 2015 Jan;8(1):46-54. doi: 10.1177/1756285614562419. [Article]
- FDA Approved Drugs: Reyvow [Link]
- AChemBlock: Lasmiditan hemisuccinate MSDS [Link]
- FDA News Release: Lasmiditan Approval [Link]
CN1CCC(CC1)C(=O)C2=NC(=CC=C2)NC(=O)C3=C(C=C(C=C3F)F)F.CN1CCC(CC1)C(=O)C2=NC(=CC=C2)NC(=O)C3=C(C=C(C=C3F)F)F.C(CC(=O)O)C(=O)O































 (what is this?) (verify)
(what is this?) (verify)