















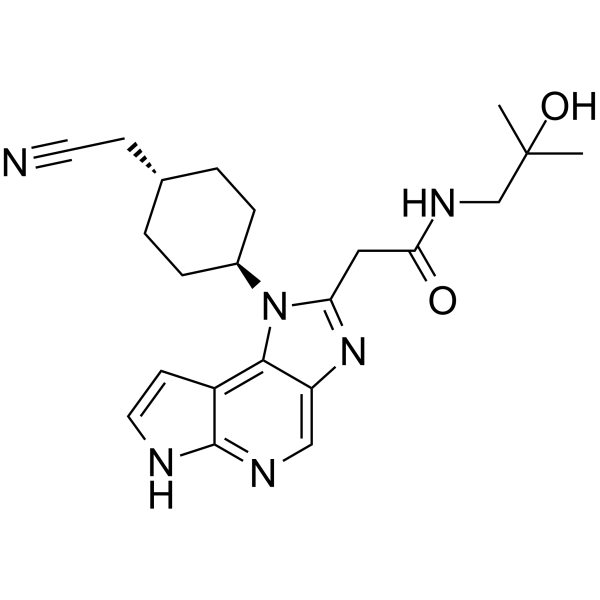
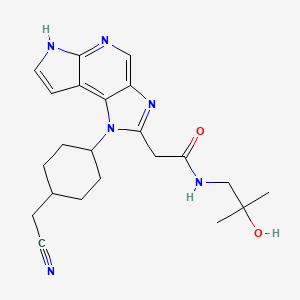
LORPUCITINIB
JNJ 64251330
2230282-02-5
UNII-OE1QTY7C25
| Molecular Weight |
408.50 |
|---|---|
| Formula |
C22H28N6O2 |
| 1-(TRANS-4-(CYANOMETHYL)CYCLOHEXYL)-1,6-DIHYDRO-N-(2-HYDROXY-2-METHYLPROPYL)IMIDAZO(4,5-D)PYRROLO(2,3-B)PYRIDINE-2-ACETAMIDE |
2-[3-[4-(cyanomethyl)cyclohexyl]-3,5,8,10-tetrazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,11-pentaen-4-yl]-N-(2-hydroxy-2-methylpropyl)acetamide
is a Gut-Restricted JAK Inhibitor for the research of Inflammatory Bowel Disease.
Lorpucitinib is an orally bioavailable pan-inhibitor of the Janus associated-kinases (JAKs), with potential immunomodulatory and anti-inflammatory activities. Upon oral administration, lorpucitinib works in the gastrointestinal (GI) tract where it targets, binds to and inhibits the activity of the JAKs, thereby disrupting JAK-signal transducer and activator of transcription (STAT) signaling pathways and the phosphorylation of STAT proteins. This may inhibit the release of pro-inflammatory cytokines and chemokines, reducing inflammatory responses and preventing inflammation-induced damage. The Janus kinase family of non-receptor tyrosine kinases, which includes tyrosine-protein kinase JAK1 (Janus kinase 1; JAK1), tyrosine-protein kinase JAK2 (Janus kinase 2; JAK2), tyrosine-protein kinase JAK3 (Janus kinase 3; JAK3) and non-receptor tyrosine-protein kinase TYK2 (tyrosine kinase 2), plays a key role in cytokine signaling and inflammaton.
PATENT
WO2019239387
WO2018112379
WO2018112382
PATENT
WO/2022/189496LORPUCITINIB FOR USE IN THE TREATMENT OF JAK MEDIATED DISORDERS
Example 1
[0117] 2-(1-((1r,4r)-4-(Cyanomethyl)cyclohexyl)-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-2-yl)-N-(2-hydroxy-2-methylpropyl)acetamide
Step A: 2-(1-((1r,4r)-4-(Cyanomethyl)cyclohexyl)-6-(phenylsulfonyl)-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-2-yl)-N-(2-hydroxy-2-methylpropyl)acetamide. To ensure dry starting material, ethyl 2-(1-((1r,4r)-4-(cyanomethyl)cyclohexyl)-6-(phenylsulfonyl)-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-2-yl)acetate (Intermediate 3) was heated under vacuum at 50 °C for 18 h prior to the reaction. In a 1 L flask, ethyl 2-(1-((1r,4r)-4-(cyanomethyl)cyclohexyl)-6-(phenylsulfonyl)-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-2-yl)acetate (Intermediate 3, 52.585 g, 104.01 mmol) was suspended in DMA (50 mL). 1-Amino-2-methylpropan-2-ol (50 mL) was added and the reaction was heated to 110 °C for 45 minutes, then to 125 °C for 5 hours. The reaction was cooled to room temperature and diluted with EtOAc (800 mL). The organic layer was extracted three times with a solution of water/ brine wherein the solution was made up of 1 L water plus 50 mL brine. The aqueous layers were back extracted with EtOAc (2 × 600 mL). The combined organic layers were dried over anhydrous MgSO4,
concentrated to dryness, and then dried for 3 days under vacuum to provide the title compound (65.9 g, 98% yield) as a yellow foam. The product was taken to the next step with no further purification. MS (ESI): mass calcd. for C28H32N6O4S, 548.22; m/z found, 549.2 [M+H]+.1H NMR (400 MHz, CDCl3): δ 8.76 (s, 1H), 8.26 – 8.19 (m, 2H), 7.84 (d, J = 4.1 Hz, 1H), 7.60 – 7.53 (m, 1H), 7.50 – 7.44 (m, 2H), 6.84 (d, J = 4.2 Hz, 1H), 4.76 – 4.61 (m, 1H), 3.97 (s, 2H), 3.45 (s, 1H), 3.27 (d, J = 5.9 Hz, 2H), 2.41 (d, J = 6.5 Hz, 2H), 2.38 – 2.25 (m, 2H), 2.23 – 2.12 (m, 2H), 2.09 -1.94 (m, 4H), 1.48 (qd, J = 13.6, 4.0 Hz, 2H), 1.21 (s, 6H).
[0118] Step B: 2-(1-((1r,4r)-4-(Cyanomethyl)cyclohexyl)-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-2-yl)-N-(2-hydroxy-2-methylpropyl)acetamide. 2-(1-((1r,4r)-4-(Cyanomethyl)cyclohexyl)-6-(phenylsulfonyl)-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-2-yl)-N-(2-hydroxy-2-methylpropyl)acetamide (65.90 g, 102.1 mmol) was added to a 1 L flask containing a stir bar. 1,4-dioxane (300 mL) was added, followed by aq KOH (3 M, 150 mL). The reaction was heated at 80 °C for 2 h. The reaction was cooled to room temperature and the solvent volume was reduced to about 200 mL on a rotovap. The residue was treated with a solution of water/brine (100 mL/100mL), then extracted with 10% MeOH in CH2Cl2 (2 x 1L). The organic layers were combined, dried over anhydrous MgSO4, and concentrated to dryness to provide a yellow solid. The solid was suspended in CH2Cl2 (200 mL), stirred vigorously for 30 minutes, and then collected by filtration. The solid was rinsed with CH2Cl2 (100 mL), dried by pulling air through the filter, and then further dried under vacuum at room temperature for 16 h to provide the title compound (41.59 g, 89% yield) as a white solid. MS (ESI): mass calcd. for C22H28N6O2, 408.23; m/z found, 409.2 [M+H]+. 1H NMR (600 MHz, DMSO-d6): δ 11.85 (s, 1H), 8.50 (s, 1H), 8.21 – 8.10 (m, 1H), 7.49 – 7.43 (m, 1H), 6.74 – 6.65 (m, 1H), 4.53 – 4.42 (m, 2H), 4.07 (s, 2H), 3.08 (d, J = 6.0 Hz, 2H), 2.58 (d, J = 6.1 Hz, 2H), 2.41 – 2.28 (m, 2H), 2.09 – 1.92 (m, 5H), 1.42 – 1.31 (m, 2H), 1.09 (s, 6H). The synthesis and active compound characterization of each of the aspects of this invention are provided herein in the form of examples. Due to the crystal structure of some of the aspects of this invention, polymorph screening may be pursued to further characterize specific forms of any such compound. This is illustrated in a non-limiting manner for compound of Formula I by the example under the heading polymorph screening.
[0119] The following compounds were prepared in reference to the foregoing synthesis:
Intermediate 1
[0120] 2-((1r,4r)-4-((5-Nitro-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)cyclohexyl)acetonitrile
[0121] Step A: tert-butyl N-[(1r,4r)-4-(Hydroxymethyl)cyclohexyl]carbamate. To a 20-L 4-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen was placed (1r,4r)-4-[[(tert-butoxy)carbonyl]amino]cyclohexane-1-carboxylic acid (1066 g, 4.38 mol, 1.00 equiv) and THF (10 L). This was followed by the dropwise addition of BH3-Me2S (10 M, 660 mL) at -10 °C over 1 h. The resulting solution was stirred for 3 h at 15 °C. This reaction was performed three times in parallel and the reaction mixtures were combined. The reaction was then quenched by the addition of methanol (2 L). The resulting mixture was concentrated under vacuum. This resulted in of tert-butyl N-[(1r,4r)-4-(hydroxymethyl)cyclohexyl]carbamate (3000 g, 99.6%) as a white solid. MS (ESI): mass calcd. for C12H23NO3, 229.32; m/z found, 215.2 [M-tBu+MeCN+H]+; 1H NMR: (300 MHz, CDCl3): δ 4.40 (s, 1H), 3.45 (d, J = 6.3 Hz, 2H), 3.38 (s, 1H), 2.05-2.02 (m, 2H), 1.84-1.81 (m, 2H), 1.44 (s, 11H), 1.17-1.01 (m, 4H).
[0122] Step B: tert-butyl N-[(1r,4r)-4-[(Methanesulfonyloxy)methyl]cyclohexyl]carbamate. To a 20 L 4-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed tert-butyl N-[(1r,4r)-4-(hydroxymethyl)cyclohexyl]carbamate (1000 g, 4.36 mol, 1.00 equiv.), dichloromethane (10 L), pyridine (1380 g, 17.5 mol, 4.00 equiv.). This was followed by the dropwise addition of MsCl (1000 g, 8.73 mol, 2.00 equiv.) at -15 °C. The resulting solution was stirred overnight at 25 °C. This reaction was performed in parallel for 3 times and the reaction mixtures were combined. The reaction was then quenched by the addition of 2 L of water. The
water phase was extracted with ethyl acetate (1 x 9 L). The organic layer was separated and washed with 1 M HCl (3 x 10 L), NaHCO3 (saturated aq.) (2 x 10 L), water (1 x 10 L) and brine (1 x 10 L). The mixture was dried over anhydrous sodium sulfate, filtered and concentrated under vacuum. This resulted in of tert-butyl N-[(1r,4r)-4-[(methanesulfonyloxy)methyl]cyclohexyl]carbamate (3300 g, 82%) as a white solid. LC-MS: MS (ESI): mass calcd. for C13H25NO5S, 307.15; m/z found 292.1, [M-tBu+MeCN+H]+; 1H NMR: (300 MHz, CDCl3): δ 4.03 (d, J = 6.6 Hz, 2H), 3.38 (s, 1H), 3.00 (s, 3H), 2.07-2.05 (m, 2H), 1.87-1.84 (m, 2H), 1.72-1.69 (m, 1H), 1.44 (s, 9H), 1.19-1.04 (m, 4H).
[0123] Step C: tert-butyl N-[(1r,4r)-4-(Cyanomethyl)cyclohexyl]carbamate. To a 10 L 4-necked round-bottom flask, was placed tert-butyl N-[(1r,4r)-4-[(methanesulfonyloxy)methyl]cyclohexyl]carbamate (1100 g, 3.58 mol, 1.00 equiv.), DMSO (5500 mL) and NaCN (406 g, 8.29 mol, 2.30 equiv.). The resulting mixture was stirred for 5 h at 90 °C. This reaction was performed in parallel 3 times and the reaction mixtures were combined. The reaction was then quenched by the addition of 15 L of water/ice. The solids were collected by filtration. The solids were washed with water (3 x 10 L). This resulted in tert-butyl N-[(1r,4r)-4-(cyanomethyl)cyclohexyl]carbamate (2480 g, 97%) as a white solid. MS (ESI): mass calcd. for C13H22N2O2, 238.17; m/z found 224 [M-tBu+MeCN+H]+; 1H NMR: (300 MHz, CDCl3): δ 4.39 (s, 1H), 3.38 (s, 1H), 2.26 (d, J = 6.9 Hz, 2H), 2.08-2.04 (m, 2H), 1.92-1.88 (m, 2H), 1.67-1.61 (m, 1H), 1.44 (s, 9H), 1.26-1.06 (m, 4H).
[0124] Step D: 2-[(1r,4r)-4-Aminocyclohexyl]acetonitrile hydrochloride. To a 10-L round-bottom flask was placed tert-butyl N-[(1r,4r)-4-(cyanomethyl)cyclohexyl]carbamate (620 g, 2.60 mol, 1.00 equiv.), and 1,4-dioxane (2 L). This was followed by the addition of a solution of HCl in 1,4-dioxane (5 L, 4 M) dropwise with stirring at 10 °C. The resulting solution was stirred overnight at 25 °C. This reaction was performed for 4 times and the reaction mixtures were combined. The solids were collected by filtration. The solids were washed with 1,4-dioxane (3 x 3 L), ethyl acetate (3 x 3 L) and hexane (3 x 3 L). This resulted in 2-[(1r,4r)-4-aminocyclohexyl]acetonitrile hydrochloride (1753 g, 96%) as a white solid. MS (ESI): mass calcd. for C8H14N2, 138.12; m/z found 139.25, [M+H]+; 1H NMR: (300 MHz, DMSO-d6): δ 8.14 (s, 3H), 2.96-2.84 (m, 1H), 2.46 (d, J = 6.3 Hz, 2H), 1.98 (d, J = 11.1 Hz, 2H), 1.79 (d, J = 12.0 Hz, 2H), 1.64-1.49 (m, 1H), 1.42-1.29 (m, 2H), 1.18-1.04 (m, 2H).
[0125] Step E: 2-((1r,4r)-4-((5-Nitro-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)cyclohexyl)acetonitrile. To a 1000 mL round bottom flask containing 2-[(1r,4r)-4-aminocyclohexyl]acetonitrile hydrochloride (29.10 g, 166.6 mmol) was added DMA (400 mL). The resulting suspension was treated with 4-chloro-5-nitro-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridine (51.53 g, 152.6 mmol), followed by DIPEA (63.0 mL, 366 mmol). The reaction mixture was placed under N2 and heated at 80 °C for 4 h. The crude reaction mixture was cooled to room temperature and slowly poured into a vigorously stirred 2 L flask containing 1.6 L water. The resulting suspension was stirred for 15 minutes at room temperature, then filtered and dried for 16 h in a vacuum oven with heating at 70 °C to provide the title compound (63.37 g, 95%) as a yellow solid. MS (ESI): mass calcd. for C21H21N5O4S, 439.1; m/z found, 440.1 [M+H]+. 1H NMR (500 MHz, CDCl3): δ 9.10 (s, 1H), 8.99 (d, J = 7.8 Hz, 1H), 8.23 – 8.15 (m, 2H), 7.66 – 7.59 (m, 2H), 7.56 – 7.49 (m, 2H), 6.67 (d, J = 4.2 Hz, 1H), 3.95 – 3.79 (m, 1H), 2.38 (d, J = 6.2 Hz, 2H), 2.32 -2.21 (m, 2H), 2.08 – 1.98 (m, 2H), 1.88 – 1.76 (m, 1H), 1.60 – 1.32 (m, 4H).
Intermediate 2
[0126] 2-((1r,4r)-4-((5-Amino-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)cyclohexyl)acetonitrile
[0127] 2-((1r,4r)-4-((5-Nitro-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)cyclohexyl)acetonitrile (Intermediate 1, 58.60 g, 133.3 mmol) was dissolved in THF/MeOH (1:1, 4800 mL). The mixture was passed through a continuous-flow hydrogenation reactor (10% Pd/C), such as a Thales Nano H-Cube®, at 10 mL/min with 100 % hydrogen (atmospheric pressure, 80 °C), then the solution was concentrated to provide the product as a purple solid. The solid was triturated with EtOAc (400 mL) and then triturated again with MeOH (200 mL) then filtered and dried under vacuum to provide the title compound (50.2 g, 91.9% yield).
MS (ESI): mass calcd. for C21H23N5O2S, 409.2; m/z found, 410.2 [M+H]+. 1H NMR (400 MHz, CDCl3) δ 8.10 – 8.03 (m, 2H), 7.76 (s, 1H), 7.51 – 7.43 (m, 1H), 7.43 – 7.34 (m, 3H), 6.44 (d, J = 4.2 Hz, 1H), 4.61 (d, J = 8.5 Hz, 1H), 3.65 – 3.51 (m, 1H), 2.74 (s, 2H), 2.26 (d, J = 6.4 Hz, 2H), 2.19 – 2.05 (m, 2H), 1.97 – 1.86 (m, 2H), 1.76 – 1.59 (m, 1H), 1.33 – 1.12 (m, 4H).
Intermediate 3
[0128] Ethyl 2-(1-((1r,4r)-4-(cyanomethyl)cyclohexyl)-6-(phenylsulfonyl)-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-2-yl)acetate
[0129] To a 1L round bottom flask containing a stir bar and 2-((1r,4r)-4-((5-amino-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)cyclohexyl)acetonitrile (Intermediate 2, 58.31 g, 142.4 mmol) was added ethyl 3-ethoxy-3-iminopropanoate (60.51 g, 309.3 mmol), followed by EtOH (600 mL, dried over 3Å molecular sieves for 48 h). A reflux condenser was attached to the reaction flask, the reaction was purged with N2, and was heated at 90 °C for 9 h. The reaction mixture was cooled to room temperature and left to stand for 30 h where the product crystallized out as brown needles. The solids were broken up with a spatula and the reaction mixture was transferred to a 2 L flask. Water (1.4 L) was added slowly via separatory funnel with vigorous stirring. After addition of the water was complete, the suspension was stirred for 30 minutes. The brown needles were isolated by filtration and then dried by pulling air through the filter for 1 h. The product was transferred to a 500 mL flask and treated with EtOAc (200 mL). A small quantity of seed crystals were added, which induced the formation of a white solid precipitate. The suspension was stirred for 30 minutes at room temperature, filtered, rinsed with EtOAc (25 mL), and dried under vacuum to provide the product as a white solid (48.65 g, 68% yield). MS (ESI): mass calcd. for C26H27N5O4S, 505.2; m/z found, 506.2 [M+H]+. 1H NMR (400
MHz, CDCl3) δ 8.85 (s, 1H), 8.28 – 8.19 (m, 2H), 7.84 (d, J = 4.0 Hz, 1H), 7.61 – 7.53 (m, 1H), 7.52 – 7.43 (m, 2H), 6.84 (d, J = 4.1 Hz, 1H), 4.32 (s, 1H), 4.20 (q, J = 7.1 Hz, 2H), 4.09 (s, 2H), 2.44 (d, J = 6.2 Hz, 2H), 2.40 – 2.27 (m, 2H), 2.16 (d, J = 13.3 Hz, 2H), 2.12 – 1.96 (m, 3H), 1.54 – 1.38 (m, 2H), 1.27 (t, J = 7.1 Hz, 3H).
Polymorph screening example
[0130] Some embodiments of compound of Formula I as free bases present multiple crystalline configurations that have a complex solid-state behavior, some of which in turn can present distinguishing features among themselves due to different amounts of incorporated solvent. Some embodiments of compound of Formula I are in the form of pseudopolymorphs, which are embodiments of the same compound that present crystal lattice compositional differences due to different amounts of solvent in the crystal lattice itself. In addition, channel solvation can also be present in some crystalline embodiments of compound of Formula I, in which solvent is incorporated within channels or voids that are present in the crystal lattice. For example, the various crystalline configurations given in Table 2 were found for compound of Formula I. Because of these features, non-stoichiometric solvates were often observed, as illustrated in Table 2. Furthermore, the presence of such channels or voids in the crystal structure of some embodiments according to this invention enables the presence of water and/or solvent molecules that are held within the crystal structure with varying degrees of bonding strength. Consequently, changes in the specific ambient conditions can readily lead to some loss or gain of water molecules and/or solvent molecules in some embodiments according to this invention. It is understood that “solvation” (third column in Table 2) for each of the embodiments listed in Table 2 is the formula solvation, and that the actual determination of the same as a stoichiometry number (fourth column in Table 2) can slightly vary from the formula solvation depending on the actual ambient conditions when it is experimentally determined. For example, if about half of the water molecules in an embodiment may be present as hydrogen-bonded to the active compound in the crystal lattice, while about the other half of water molecules may be in channels or voids in the crystal lattice, then changes in ambient conditions may alter the amount of such loosely contained water molecules in voids or channels, and hence lead to a slight difference between the formula solvation that is assigned according to, for example, single crystal diffraction, and the
stoichiometry that is determined by, for example, thermogravimetric analysis coupled with mass spectroscopy.
Table 2. Embodiments of crystalline forms of compound of Formula I
[0131] The compound that was obtained as described in Example 1 was further crystallized by preparing a slurry in DCM (1:3, for example 10 g of compound in 30 ml DCM) that was stirred at 40oC for 4 hours, and further stirred for 14 hours at 25oC, then heptane was slowly added (1:2, for example 20 ml of heptane into the compound/DCM slurry/solution) at 25oC, stirred at 40oC for 4 hours, cooled to 25oC and stirred for further 14 hours at 25oC. Subsequent filtration led to compound of Formula I in the form of an off-white solid, that was identified as a monohydrate, a 1s embodiment.
CLIP
Journal of Medicinal Chemistry (2020), 63(6), 2915-2929

/////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Clip
https://clinicaltrials.gov/ct2/show/NCT04552197
The purpose of this study is to evaluate: systemic and local gut (rectum and sigmoid colon) exposure to JNJ-64251330, local tissue Pharmacodynamics (PD) using gut (rectum and sigmoid colon) biopsies (Part 1) and the effect of food on the rate and extent of absorption of JNJ-64251330 from oral tablet dosed with or without food (Part 2).
Familial adenomatous polyposis (FAP) is the most common polyposis syndrome. It is an autosomal dominant inherited disorder characterized by the early onset of hundreds to thousands of adenomatous polyps throughout the colon. JNJ-64251330 (lorpucitinib) is an oral, small molecule, potent pan-janus kinase (JAK) inhibitor that blocks phosphorylation of Signal Transducer and Activator of Transcription (STAT) proteins. pSTAT induces transcription of multiple genes involved in the progression of inflammatory disease. JNJ-64251330 has chemical properties that limits the amount of drug in the blood while delivering the drug to the tissues of the gut. Local inhibition of JAK in the gut may present a promising method to treat inflammatory diseases of the intestinal tract, such as FAP. The study consists of 3 phases: screening phase (30 days) a treatment phase (24 weeks), and follow-up visit (up to 30 days after last dose of study drug). The total duration of the study will be up to 32 weeks. Study evaluations will include efficacy via endoscopies, safety (monitoring of adverse events (AE), serious adverse events (SAEs), events of infections including tuberculosis (TB), clinical laboratory blood tests (complete blood count and serum chemistries), vital signs, and concomitant medication review), pharmacokinetics, pharmacodynamic and biomarkers evaluations.
Adenomatous polyposis coli (APC) also known as deleted in polyposis 2.5 (DP2.5) is a protein that in humans is encoded by the APC gene.[4] The APC protein is a negative regulator that controls beta-catenin concentrations and interacts with E-cadherin, which are involved in cell adhesion. Mutations in the APC gene may result in colorectal cancer.[5]
APC is classified as a tumor suppressor gene. Tumor suppressor genes prevent the uncontrolled growth of cells that may result in cancerous tumors. The protein made by the APC gene plays a critical role in several cellular processes that determine whether a cell may develop into a tumor. The APC protein helps control how often a cell divides, how it attaches to other cells within a tissue, how the cell polarizes and the morphogenesis of the 3D structures,[6] or whether a cell moves within or away from tissue. This protein also helps ensure that the chromosome number in cells produced through cell division is correct. The APC protein accomplishes these tasks mainly through association with other proteins, especially those that are involved in cell attachment and signaling. The activity of one protein in particular, beta-catenin, is controlled by the APC protein (see: Wnt signaling pathway). Regulation of beta-catenin prevents genes that stimulate cell division from being turned on too often and prevents cell overgrowth.
The human APC gene is located on the long (q) arm of chromosome 5 in band q22.2 (5q22.2). The APC gene has been shown to contain an internal ribosome entry site. APC orthologs[7] have also been identified in all mammals for which complete genome data are available.
////////////////JNJ-64251330, JNJ 64251330, LORPUCITINIB, PHASE 1, CANCER, Adenomatous Polyposis Coli
O=C(NCC(C)(O)C)CC1=NC2=CN=C(NC=C3)C3=C2N1[C@H]4CC[C@H](CC#N)CC4