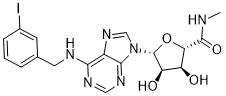

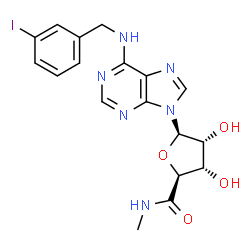
CF 101, Piclidenoson
ALB-7208
CAS 152918-18-8
Chemical Formula: C18H19IN6O4
Molecular Weight: 510.28
(2S,3S,4R,5R)-3,4-Dihydroxy-5-{6-[(3-iodobenzyl)amino]-9H-purin-9-yl}-N-methyltetrahydro-2-furancarboxamide
N6-(3-Iodobenzyl)adenosine-5′-N-methyluronamide
β-D-Ribofuranuronamide, 1-deoxy-1-[6-[[(3-iodophenyl)methyl]amino]-9H-purin-9-yl]-N-methyl-
1-Deoxy-1-[6-[[(3-iodophenyl)methyl]amino]-9H-purin-9-yl]-N-methyl-β-D-ribofuranuronamide
10136
1-Deoxy-1-[6-[((3-Iodophenyl)methyl)amino]-9H-purin-9-yl]-N-methyl-β-D-ribofuranuronamide
30679UMI0N
UNII-30679UMI0N
Пиклиденозон [Russian] [INN]
بيكليدينوسون [Arabic] [INN]
CF 101 (known generically as IB-MECA) is an anti-inflammatory drug for rheumatoid arthritis patients. Its novel mechanism of action relies on antagonism of adenoside A3 receptors. CF101 is supplied as an oral drug and has an excellent safety profile. It is also being considered for the treatment of other autoimmune-inflammatory disorders, such as Crohn’s disease, psorasis and dry eye syndrome.

- Originator Can-Fite BioPharma
- Class Amides; Anti-inflammatories; Antineoplastics; Antipsoriatics; Antirheumatics; Eye disorder therapies; Iodobenzenes; Neuroprotectants; Purine nucleosides; Ribonucleosides; Small molecules
- Mechanism of Action Adenosine A3 receptor agonists; Immunosuppressants; Interleukin 23 inhibitors; Interleukin-17 inhibitors
- Phase III Plaque psoriasis; Rheumatoid arthritis
- Phase II Glaucoma; Ocular hypertension
- Phase I Uveitis
- Preclinical Osteoarthritis
- Discontinued Colorectal cancer; Dry eyes; Solid tumours
- 05 Feb 2019 Can-Fite BioPharma receives patent allowance for A3 adenosine receptor (A3AR) agonists in USA
- 05 Feb 2019 Can-Fite BioPharma receives patent allowance for A3 adenosine receptor (A3AR) agonists in North America, South America, Europe and Asia
- 21 Aug 2018 Phase-III clinical trials in Plaque psoriasis (Monotherapy) in Israel (PO)
Piclidenoson, also known as CF101, is a specific agonist to the A3 adenosine receptor, which inhibits the development of colon carcinoma growth in cell cultures and xenograft murine models. CF101 has been shown to downregulate PKB/Akt and NF-κB protein expression level. CF101 potentiates the cytotoxic effect of 5-FU, thus preventing drug resistance. The myeloprotective effect of CF101 suggests its development as an add-on treatment to 5-FU.
Piclidenoson is known to be a TNF-α synthesis inhibitor and a neuroprotectant. use as an A3 adenosine receptor agonist, useful for treating rheumatoid arthritis (RA), psoriasis, osteoarthritis and glaucoma.
Can-Fite BioPharma , under license from the National Institutes of Health (NIH), is developing a tablet formulation of CF-101, an adenosine A3 receptor-targeting, TNF alpha-suppressing low molecular weight molecule for the potential treatment of psoriasis, RA and liver cancer. The company is also investigating a capsule formulation of apoptosis-inducing namodenoson, the lead from a program of adenosine A3 receptor agonist, for treating liver diseases, including hepatocellular carcinoma (HCC). In January 2019, preclinical data for the treatment of obesity were reported. Also, see WO2019105217 , WO2019105359 and WO2019105082 , published alongside.
PATENT
WO-2019105388
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019105388&tab=FULLTEXT&maxRec=1000
Novel crystalline forms of CF-101 (also known as piclidenoson; designated as Forms CS1, CS2 and CS3), processes for their preparation, compositions comprising them and their use as an A3 adenosine receptor agonist for treating rheumatoid arthritis, psoriasis, osteoarthritis and glaucoma are claimed
CF-101 was developed by Kan-Fete Biomedical Co., Ltd. By the end of 2018, CF-101 is in clinical phase III for the treatment of autoimmune diseases such as rheumatoid arthritis, osteoarthritis and psoriasis, as well as glaucoma. CF-101 is an A3 adenosine receptor (A3AR) agonist, and adenosine plays an important role in limiting inflammation through its receptor. Adenosine can produce anti-inflammatory effects by inhibiting TNF-a, interleukin-1, and interleukin-6. Studies have shown that A3AR agonists are in different experimental autoimmune models, such as rheumatoid arthritis, Crohn’s disease, and silver swarf. In the disease, it acts as an anti-inflammatory agent by improving the inflammatory process.
The chemical name of CF-101 is: 1-deoxy-I-(6-{[(3-iodophenyl)methyl]amino}-9H-fluoren-9-yl)-N-methyl-bD-ribofuranose Carbonamide (hereinafter referred to as “Compound I”) has the following structural formula:
A crystal form is a solid in which a compound molecule is orderedly arranged in a microstructure to form a crystal lattice, and a drug polymorphism phenomenon means that two or more different crystal forms of a drug exist.
Due to different physical and chemical properties, different crystal forms of drugs may have different dissolution and absorption in the body, which may affect the clinical efficacy and safety of the drug to a certain extent; especially for poorly soluble solid drugs, the crystal form will have greater influence. Therefore, the drug crystal form is inevitably an important part of drug research and an important part of drug quality control. Most importantly, the study of crystal forms is beneficial to find a crystal form that is clinically therapeutically meaningful and has stable and physicochemical properties.
There are no reports of CF-101 related crystal forms so far. Amorphous is generally not suitable as a medicinal form, and the molecules in the amorphous material are disorderly arranged, so they are in a thermodynamically unstable state. Amorphous solids are in a high-energy state, and generally have poor stability. During the production and storage process, amorphous drugs are prone to crystal transformation, which leads to the loss of consistency in drug bioavailability, dissolution rate, etc., resulting in changes in the clinical efficacy of the drug. In addition, the amorphous preparation is usually a rapid kinetic solid precipitation process, which easily leads to excessive residual solvents, and its particle properties are difficult to control by the process, making it a challenge in the practical application of the drug.
Therefore, there is a need to develop a crystalline form of CF-101 that provides a usable solid form for drug development. The inventors of the present application have unexpectedly discovered the crystalline forms CS1, CS2 and CS3 of Compound I, which have melting point, solubility, wettability, purification, stability, adhesion, compressibility, fluidity, dissolution in vitro and in vivo, and biological effectiveness. There is an advantage in at least one of the properties and formulation processing properties. Crystalline CS1 has advantages in physical and chemical properties, especially physical and chemical stability, low wettability, good solubility and good mechanical stability. It provides a new and better choice for the development of drugs containing CF-101, which is very important. The meaning.
Figure 7 is a 1 H NMR spectrum of the crystalline form CS3 obtained according to Example 7 of the present invention
The nuclear magnetic data of the crystalline form CS3 obtained in Example 7 was: { 1 H NMR (400 MHz, DMSO) δ 8.82 – 8.93 (m, 1H), 8.53 – 8.67 (m, 1H), 8.45 (s, 1H), 8.31 ( s, 1H), 7.73 (s, 1H), 7.59 (d, J = 7.7 Hz, 1H), 7.36 (d, J = 7.7 Hz, 1H), 7.11 (t, J = 7.8 Hz, 1H), 5.98 ( d, J = 7.4 Hz, 1H), 5.74 (s, 1H), 5.56 (s, 1H), 4.64 (d, J = 29.3 Hz, 3H), 4.32 (s, 1H), 4.15 (s, 1H), 2.71 (d, J = 4.6 Hz, 3H), 1.91 (s, 3H).}. Form CS3 has a single peak at 1.91, corresponding to the hydrogen chemical shift of the acetic acid molecule. According to the nuclear magnetic data, the molar ratio of acetic acid molecule to CF-101 is 1:1, and its 1 H NMR is shown in FIG.7
PAPER
Journal of medicinal chemistry (1994), 37(5), 636-46
https://pubs.acs.org/doi/pdf/10.1021/jm00031a014
PAPER
Journal of medicinal chemistry (1998), 41(10), 1708-15
https://pubs.acs.org/doi/abs/10.1021/jm9707737
PAPER
Bioorganic & Medicinal Chemistry (2006), 14(5), 1618-1629
PATENT
WO 2015009008
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015009008
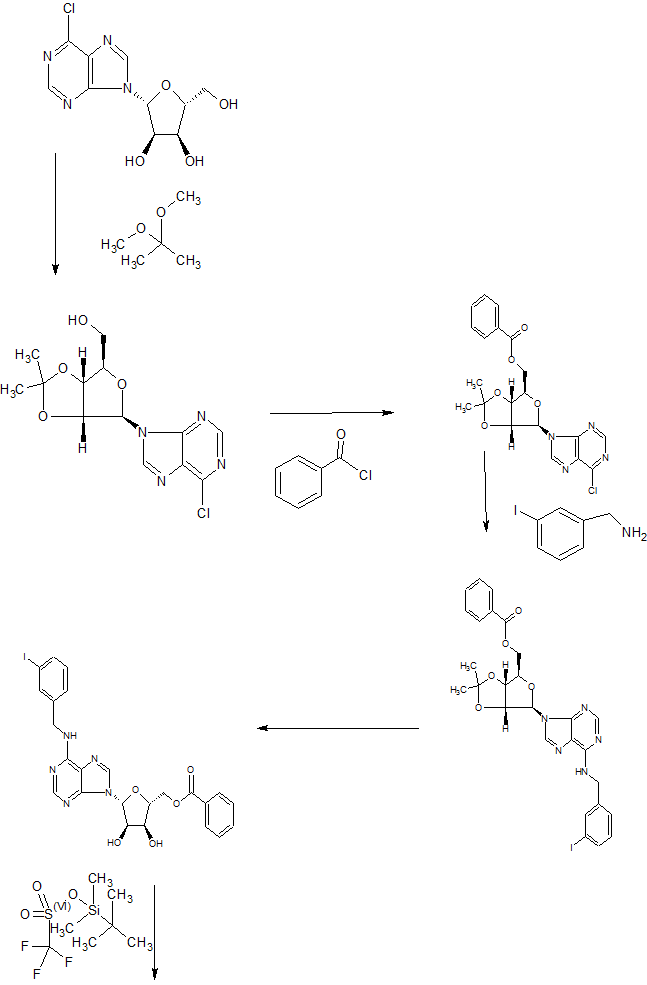
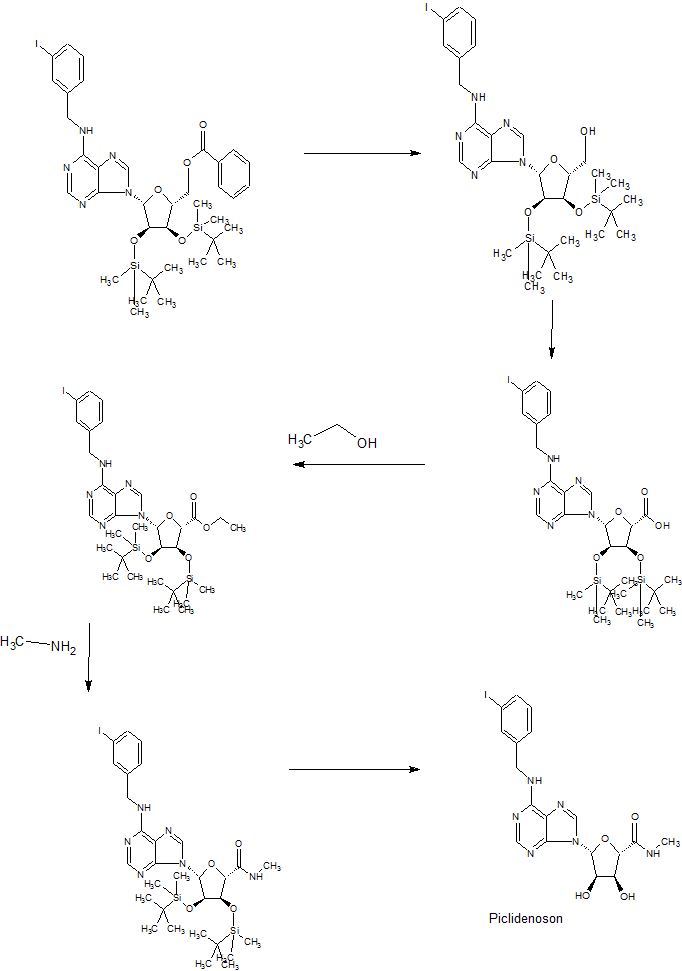



Preparation Example 1: Synthesis of Compound (5) (S) -2 – ((R) -1- (2-Chloro-6- (3-iodobenzylamino) -9H- purin- Hydroxyethoxy) -3-hydroxy-N-methylpropanamide)
Step 1: A solution of (2R, 3S, 4S, 5R) -2- (benzoyloxymethyl) -5- (2,6- dichloro-9H- purin-9- yl) tetrahydrofuran- Preparation of benzoate (7)
Starting material A mixture of (2R, 3R, 4S, 5R) -2-acetoxy-5- (benzoyloxymethyl) tetrahydrofuran-3,4-diyldibenzoate (7.5 g, 14.9 mmol) (3.09 g, 16.4 mmol) was dissolved in acetonitrile (50 mL), and a solution of N, O-bis (trimethylsilyl) acetamid (8.9 mL, 36.4 mmol) was slowly added dropwise for 10-15 minutes Then, the mixture is stirred at 60 DEG C for 30 minutes. After cooling the reaction solution to -30 ° C, TiCl 4 (60 mL, 1 M methylene chloride solution, 59.5 mmol) is added dropwise, and the mixture is stirred at 60-65 ° C for 20 minutes. After confirming the completion of the reaction, methylene chloride (500 mL) and saturated sodium hydrogencarbonate solution (500 mL) are added. The reaction solution was stirred at 0 ° C for 30 minutes, and the organic layer was extracted and dried over anhydrous magnesium sulfate. After concentration under reduced pressure, the obtained residue was separated by column chromatography to obtain the intermediate compound (2R, 3S, 4S, 5R) -2- (benzoyloxymethyl) -5- (2,6- dichloro- Yl) tetrahydrofuran-3,4-diyl dibenzoate (9.3 g, 98.8%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; CDCl 3 ) δ ppm 4.71-4.74 (dd, J = 12.22, 3.91 Hz, 1H), 4.85-4.93 (m, 2H), 6.12-6.14 (t, J = 4.89 Hz, 1H) (M, 1H), 6.16-6.19 (t, J = 5.38 Hz, 1H), 6.47-6.48 (d, J = 5.38 Hz, 1H), 7.35-7.38 4H), 7.54-7.61 (m, 3H), 7.92-7.93 (d, J = 7.33 Hz, 2H), 8.02-8.06 (m, 4H), 8.28 (s, 1H); 13C NMR (125 MHz; CDCl 3 ) δ 63.50, 71.59, 74.33, 81.56, 87.05, 128.14, 128.59 (3), 128.63 (2), 128.73 (2), 129.10, 129.63 (2), 129.88 (2), 129.92 (2), 131.38, 133.63, 133.87, 133.98, 143.81, 152.36, 152.64, 153.51, 165.13, 165.29, 166.03; mp = 76-80 [deg.] C.
Step 2: (2R, 3S, 4S, 5R) -2- (Benzoyloxymethyl) -5- (2-chloro-6- (3-iodobenzylamino) -9H- purin-9-yl) tetrahydrofuran -3,4-diyl dibenzoate (8)
The intermediate compound (204 mg, 0.32 mmol) and 3-iodobenzylamine hydrochloride (113 mg, 0.41 mmol) prepared in the above step 1 were dissolved in anhydrous ethanol (5 mL) under a nitrogen atmosphere, triethylamine (0.13 mL, 0.96 mmol) is stirred at room temperature for 24 hours. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure, and the obtained residue was separated by column chromatography to obtain the intermediate compound (2R, 3S, 4S, 5R) -2- (benzoyloxymethyl) (3-iodobenzylamino) -9H-purin-9-yl) tetrahydrofuran-3,4-diyldibenzoate (230 mg, 86.14%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; CDCl 3 ) δ ppm 4.71-4.90 (m, 5H), 6.13-6.17 (m, 2H), 6.33 (.. Br s, 1H), 6.43-6.44 (d, J = 4.89 Hz (M, 4H), 7.31-7.33 (m, 6H), 7.33-7.46 (m, 6H) 7.70-7.71 (t, J = 1.46 Hz, 1H), 7.88 (s, 1H), 7.94-7.96 (m, 2H), 7.99-8.01 (m, 2H), 8.07-8.09 (m, 2H); 13 C NMR (125 MHz; CDCl 3) δ 43.91, 63.79, 71.56, 74.43, 80.97, 86.40, 94.49, 119.07, 127.14, 128.40, 128.52 (3), 128.62 (2), 128.71, 129.32, 129.68 (3), 129.86 (2), 129.93 (2), 130.39 (2), 133.41, 133.69, 133.78, 136.72, 136.80, 138.39, 140.20, 150.05, 155.00, 165.17, 165.31, 166.11; mp = 80-84 [deg.] C.
Step 3: ((3aR, 4R, 6R, 6aR) -6- (2-Chloro-6- (3-iodobenzylamino) -9H- purin-9- yl) -2,2- dimethyltetrahydrofur [3,4-d] [1,3] dioxol-4-yl) methanol (9)
The intermediate compound (20 g, 24.09 mmol) prepared in the above step 2 was dissolved in methanolic ammonia (1 L) and stirred at room temperature for 3 days. The reaction mixture was concentrated under reduced pressure to obtain a triol intermediate. The triol intermediate thus obtained (20 g, 38.63 mmol) was dissolved in anhydrous acetone (400 mL), and 2,2-dimethoxypropane (23.68 mL, 193.15 mmol) and p-toluenesulfonic acid monohydrate (7.34 g, 38.63 mmol) was added dropwise thereto, followed by stirring at room temperature for 12 hours. After confirming the completion of the reaction, saturated sodium hydrogencarbonate solution (400 mL) was added thereto. The reaction mixture was concentrated under reduced pressure. The organic layer was extracted with chloroform (4 x 250 mL), washed with a saturated aqueous sodium chloride solution and dried over anhydrous magnesium sulfate. The reaction mixture was concentrated under reduced pressure, and the obtained residue was then separated by column chromatography to obtain the intermediate compound ((3aR, 4R, 6R, 6aR) -6- (2-Chloro-6- (3-iodobenzylamino) Yl] -2,2-dimethyltetrahydrofuro [3,4-d] [1,3] dioxol-4-yl) methanol (12 g, 89.35%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; CDCl 3 )? Ppm 1.36 (s, 3H), 1.62 (s, 3H), 3.77-3.80 (dd, J = 12.71, 1.95 Hz, 1H), 3.95-3.98 12.71, 1.95 Hz, 1H), 4.48-4.49 (d, J = 1.46 Hz, 1H), 4.68 (br. s., 1H, exchangeable with d 2 O, OH), 4.74 (br. s., 2H), (D, J = 5.86, 1.46 Hz, 1H), 5.15-5.17 (t, J = 5.38 Hz, 1H), 5.77-5.78 (d, J = 4.40 Hz, 1H), 6.81 (br s. , 1H, exchangeable with d 2 O, NH), 7.03-7.06 (t, J = 7.82 Hz, 1H), 7.30-7.31 (d, J = 7.33 Hz, 1H), 7.59-7.61 (d, J = 7.82 Hz , & Lt; / RTI & gt; 1H), 7.67 (s, 1H), 7.70 (s, 1H); 13 C NMR (125 MHz; CDCl 3) [delta] 25.26, 27.63, 43.93, 63.37, 81.52, 82.98, 86.12, 93.89, 94.55, 114.18, 120.09, 127.19, 130.44, 136.86 (2), 139.93, 140.01, 148.80, 154.50, 155.14; mp = 82-86 [deg.] C.
Step 4: (2S, 5R) -5- (2-Chloro-6- (3-iodobenzylamino) -9H- purin-9- yl) -3,4- dihydroxy- -2-carboxamide & lt; / RTI & gt; (10)
The intermediate compound (15 g, 26.89 mmol) prepared in step 3 was dissolved in a solution of acetonitrile-water (130 mL, 1: 1) and then (diacetoxy iodo) -benzene (19 g, 59.16 mmol) 2,2,6,6-Tetramethyl 1-piperidinyloxyl (840 mg, 5.37 mmol) was added dropwise, followed by stirring at room temperature for 4 hours. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure to obtain an acid intermediate without purification. The obtained intermediate (15 g, 26.23 mmol) was dissolved in anhydrous ethanol (500 mL) under a nitrogen stream, cooled to 0 ° C, thionyl chloride (9.52 mL, 131.17 mmol) was slowly added dropwise and the mixture was stirred at room temperature for 12 hours. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure to obtain an ethyl ester intermediate without purification. Methylamine (750 mL, 2 N THF solution) was added dropwise to the resulting ethyl ester intermediate (15.5 g, 25.84 mmol) and the mixture was stirred at room temperature for 12 hours. After confirming the completion of the reaction, the reaction mixture was concentrated under reduced pressure. The obtained residue was purified by column chromatography to obtain the intermediate compound (2S, 5R) -5- (2-Chloro-6- (3- -9-yl) -3,4-dihydroxy-N-methyltetrahydrofuran-2-carboxamide (5 g, 31.80%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; DMSO-d 6 )? Ppm 2.72-2.73 (d, J = 3.91 Hz, 3H), 4.17 (brs, 1H), 4.33 (s, 1H), 4.55-4.56 (D, J = 6.35 Hz, 1H, exchangeable with D 2 O, 2′-OH), 5.71-5.72 d, J = 3.91 Hz, 1H, exchangeable with d 2 O, 3′-OH), 5.92-5.93 (d, J = 7.33 Hz, 1H), 7.11-7.14 (t, J = 7.82 Hz, 1H), 7.35 (D, J = 6.84 Hz, 1H), 7.59-7.61 (d, J = 7.82 Hz, 1H), 7.75 (s, 1H), 8.27-8.28 exchangeable with D 2 O, NH), 8.48 (s, 1 H), 8.98-8.99 (br. t, J = 5.86 Hz, 1H, exchangeable with D 2 O, N 6 H); 13 C NMR (125 MHz; DMSO-d 6) [delta] 26.07, 43.02, 72.79, 73.42, 84.95, 88.10, 95.12, 119.46, 127.31, 130.99, 136.06, 136.51, 141.57, 142.23, 150.00, 153.44, 155.31, 170.14; mp = 207-209 [deg.] C.
Step 5: (S) -2 – ((R) -1- (2-Chloro-6- (3-iodobenzylamino) -9H- purin-9- yl) -2-hydroxyethoxy) -3 – & lt; / RTI & gt; hydroxy-N-methylpropanamide (5)
The intermediate compound (2.0 g, 3.67 mmol) prepared in step 4 was dissolved in water / methanol (210 mL, 1: 2), cooled to 0 ° C and then sodium per iodate (1.57 g, 7.34 mmol) And then stirred at the same temperature for 2 hours. After completion of the reaction was confirmed, sodium borohydride (694 mg, 18.35 mmol) was added and stirred for 1 hour. After confirming the completion of the reaction, the reaction mixture was concentrated under reduced pressure, and the residue was concentrated under reduced pressure using toluene (3 x 50 mL). The residue was separated by column chromatography to obtain the title compound (1.58 g, 79%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; DMSO-d 6 ) δ ppm 2.40 (.. Br s, 3H), 3.53-3.60 (m, 1H), 3.71-3.73 (d, J = 10.27 Hz, 1H), 3.85 (br . s., 1H), 3.95 (br. s., 2H), 4.60 (br. s., 2H), 4.98-5.00 (t, J = 5.38 Hz, 1H, exchangeable with D 2 O, OH), 5.19 -5.20 (t, J = 5.86 Hz, 1H, exchangeable with D 2 O, OH), 5.78-5.80 (t, J = 5.38 Hz, 1H), 7.10-7.13 (t, J = 7.82Hz, 1H), 7.35 -7.36 (d, J = 6.84Hz, 1H), 7.59-7.60 (d, J = 5.86 Hz, 2H, exchangeable with D 2 O, NH), 7.73 (br. s., 1H), 8.30 (s, 1H ), 8.85 (br s, 1H, exchangeable with D 2 O, NH); 13 C NMR (125 MHz; DMSO-d 6) [delta] 25.59, 42.98, 62.02, 62.25, 80.43, 84.90, 95.11, 118.57, 127.27, 130.96, 136.03, 136.45, 140.78, 142.34, 150.69, 153.53, 155.17, 169.31; HRMS (FAB) m / z calcd for C 18 H20 ClIN 6 O 4 [M + Na] + 546.0279, found 569.0162; mp = 226-229 [deg.] C.
Preparation Example 2: Synthesis of Compound (11) ((R) -2- (1- (2-Chloro-6- (3-iodobenzylamino) -9H- purin-9-yl) -2- hydroxyethoxy) Propane-1,3-diol)
The intermediate compound (230 mg, 0.27 mmol) prepared in Step 2 of Example 1 was dissolved in methanolic ammonia (25 mL) and stirred at room temperature for 3 days. The reaction mixture was concentrated under reduced pressure to obtain a triol intermediate. The obtained triol intermediate (248 mg, 0.47 mmol) was treated in the same manner as in Step 5 of Example 1 to obtain the desired compound (109 mg, 75.69%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; DMSO-d 6 )? Ppm 3.13-3.17 (m, 1H), 3.22-3.26 (m, 1H), 3.43-3.47 (m, 2H), 3.54-3.56 (Br s, 2H), 4.41-4.42 (br t, J = 5.38 Hz, 1H, exchangeable with D 2 O, OH), 4.60 , J = 5.38 Hz, 1H, exchangeable with D 2 O, OH), 5.13 (br. s., 1H, exchangeable with D 2 O, OH), 5.80-5.82 (t, J = 4.89 Hz, 1H), 7.11 (D, J = 7.33 Hz, 1H), 7.36-7.37 (d, J = 7.33 Hz, 1H), 7.59-7.60 s, 1 H), 8.82 (br s, 1H, exchangeable with D 2 O, NH); 13 C NMR (125 MHz; DMSO-d 6) [delta] 43.01, 61.12, 61.23, 62.64, 80.90, 84.53, 95.12, 118.56, 127.36, 130.99, 136.04, 136.58, 140.68, 142.44, 150.73, 153.40, 155.12; HRMS (FAB) m / z calcd for C 17 H 19 ClIN 5 O 4 [M + Na] + 519.0170, found 542.0054; mp = 170-172 [deg.] C.
Preparation Example 3: Synthesis of Compound (12) ((S) -3-Hydroxy-2 – ((R) -2-hydroxy- 1- (6- (3-iodobenzylamino) -9H- Yl) ethoxy) -N-methylpropanamide & lt; / RTI & gt;
Step 1: ((3aR, 4R, 6R, 6aR) -6- (6-Chloro-9H- purin-9- yl) -2,2- dimethyltetrahydrofuro [3,4 d] [1,3 ] Dioxol-4-yl) methanol (14)
(Hydroxymethyl) tetrahydrofuran-3,4-diol (4.8 g, 16.74 mmol) and 2,2 & lt; RTI ID = 0.0 & -Dimethoxypropane (10.26 mL, 83.71 mmol) was dissolved in anhydrous acetone (120 mL) under a nitrogen stream, p-toluenesulfonic acid monohydrate (3.18 g, 16.74 mmol) was added dropwise and the mixture was stirred at room temperature for 4 hours . After confirming the completion of the reaction, the reaction is terminated with a saturated sodium hydrogencarbonate solution. The reaction solution was concentrated under reduced pressure, and the organic layer was extracted with chloroform (4 x 20 mL), washed with a saturated aqueous sodium chloride solution and dried over anhydrous magnesium sulfate. After concentration under reduced pressure, the obtained residue was separated by column chromatography to obtain the intermediate compound ((3aR, 4R, 6R, 6aR) -6- (6-Chloro-9H-purin-9- 3,4-d] [1,3] dioxol-4-yl) methanol (4.87 g, 89.03%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; CDCl 3 )? Ppm 1.38 (s, 3H), 1.65 (s, 3H), 3.80-3.83 (dd, J = 12.22, 1.46 Hz, 1H), 3.95-3.98 (Dd, J = 5.86, 1.46 Hz, 1H), 5.19 (d, J = 8.6 Hz, 1H), 4.53-4.55 5.21 (dd, J = 5.86, 4.40 Hz, 1H), 5.99-6.00 (d, J = 4.89 Hz, 1H), 8.25 (s, 1H), 8.75 (s, 1H); 13 C NMR (125 MHz; CDCl 3 )? 25.22, 27.55, 63.22, 81.51, 83.35, 86.43, 94.02, 114.51, 133.25, 144.73, 150.50, 151.71, 152.31; mp = 146-150 [deg.] C.
Step 2: ((3aR, 4R, 6R, 6aR) -6- (6-Chloro-9H-purin-9- yl) -2,2- dimethyltetrahydrofuro [3,4- d] 3] dioxol-4-yl) methyl benzoate (15)
The intermediate compound (2.8 g, 8.56 mmol) prepared in Step 1 was dissolved in anhydrous methylene chloride (100 mL), and then cooled to 0 ° C. Triethylamine (3.6 mL, 25.70 mmol) and dimethylaminopyridine (21 mg, 0.17 mmol). Benzoyl chloride (1.5 mL, 12.85 mmol) is slowly added dropwise at the same temperature and then stirred at room temperature for 2 hours. After confirming the completion of the reaction, the reaction is terminated with a saturated sodium hydrogencarbonate solution. The reaction solution was concentrated under reduced pressure, the organic layer was extracted with methylene chloride, washed with a saturated aqueous sodium chloride solution and dried over anhydrous magnesium sulfate. After concentration under reduced pressure, the obtained residue was separated by column chromatography to obtain the intermediate compound ((3aR, 4R, 6R, 6aR) -6- (6-Chloro-9H-purin-9- 3,4-d] [1,3] dioxol-4-yl) methyl benzoate (3.68 g, 99.72%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; CDCl 3 ) δ ppm 1.40 (s, 3H), 1.62 (s, 3H), 4.42-4.46 (dd, J = 11.73, 3.9 Hz, 1H), 4.61-4.64 (m, 2H) (D, J = 7.33 Hz, 2H), 5.51-5.13 (d, J = 2.93 Hz, 1H), 5.53-5.54 7.47-7.50 (t, J = 7.33 Hz, 1 H), 7.79-7.81 (d, J = 7.82 Hz, 2H), 8.21 (s, 1H), 8.64 (s, 1H); 13 C NMR (125 MHz; CDCl 3 ) δ 25.36, 27.15, 63.99, 81.42, 84.07, 85.04, 91.87, 114.92, 128.31 (2), 129.07, 129.39 (2), 132.42, 133.33, 144.10, 150.79, 151.40, 152.02 , 165.80; mp = 50-54 [deg.] C.
Step 3: ((3aR, 4R, 6R, 6aR) -6- (6- (3-Iodobenzylamino) -9H- purin- 9-yl) -2,2 dimethyltetrahydrofuro [3,4 -d] [1,3] dioxol-4-yl) methyl benzoate (16)
The intermediate compound (1.24 g, 2.87 mmol) prepared in the above step 2 was prepared in the same manner as in step 2 of Example 1 to give the intermediate compound ((3aR, 4R, 6R, 6aR) -6- (6- Yl) -2,2-dimethyltetrahydrofuro [3,4-d] [1,3] dioxol-4-yl) methyl benzoate (1.73 g, 96.11 %).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; CDCl 3 )? Ppm 1.42 (s, 3H), 1.63 (s, 3H), 4.45-4.59 (m, 1H), 4.59-4.61 (m, 2H), 4.80 (br s. J = 5.38 Hz, 1H), 5.17 (d, J = 3.43 Hz, 1H), 5.58-5.59 , 7.32-7.05 (t, J = 7.33 Hz, 2H), 7.49-7.52 (t, J = = 7.33 Hz, 1 H), 7.58-7.60 (d, J = 7.33 Hz, 1 H), 7.71 (s, (br. s., 1 H); 13 C NMR (125 MHz; CDCl 3 ) δ 25.47, 27.21, 43.81, 64.35, 81.71, 84.21, 85.03, 91.38, 94.55, 114.61, 120.52, 126.85, 128.32 (3), 129.43, 129.62 (2), 130.34, 133.18 , 136.53, 139.16, 140.99, 148.68, 153.34, 154.60, 166.02; mp = 68-72 [deg.] C.
Step 4: ((2R, 3R, 4R, 5R) -3,4-Bis (tert- butyldimethylsilyloxy) -5- (6- (3- iodobenzylamino) -9H-purin- ) Tetrahydrofuran-2-yl) methyl benzoate (17)
The intermediate compound (4.93 g, 7.85 mmol) prepared in the above step 3 was dissolved in 80% acetic acid (250 mL), and the mixture was refluxed at 100 ° C for 12 hours. After completion of the reaction was confirmed, the reaction solution was concentrated under reduced pressure, toluene (4 x 50 mL) was added, and the filtrate was concentrated under reduced pressure to obtain a diol intermediate without purification. The obtained diol intermediate (8.5 g, 14.47 mmol) was dissolved in anhydrous pyridine (250 mL), followed by addition of tetrabutyldimethylsilyl triflate (TBDMSOTf) (13.3 mL, 57.88 mmol) followed by stirring at 50 ° C for 5 hours. After confirming the completion of the reaction, the reaction solution was partitioned into methylene chloride / water. The organic layer was washed with water, saturated sodium hydrogencarbonate solution and saturated saturated sodium bicarbonate solution, and then dried over anhydrous magnesium sulfate. After concentration under reduced pressure, the obtained residue was separated by column chromatography to obtain the intermediate compound ((2R, 3R, 4R, 5R) -3,4-bis (tert-butyldimethylsilyloxy) -9H-purin-9-yl) tetrahydrofuran-2-yl) methylbenzoate (4.47 g, 69.73%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; CDCl 3 ) δ ppm -0.17 (s, 3H), 0.01 (s, 3H), 0.1 (s, 3H), 0.12 (s, 3H), 0.83 (s, 9H), 0.93 ( (t, J = 4.40 Hz, 1H), 4.74-4.78 (dd, J = J = 4.40 Hz, 1H), 4.82 (br s, 2H), 5.07-5.09 (t, J = 4.40 Hz, 1H), 5.88-5.89 (d, J = 7.82 Hz, 1H), 7.31-7.33 (d, J = 7.82 Hz, 1H), 7.38-7.41 (t, J = 7.82 Hz, 1H) , 7.52-7.55 (t, J = 7.82 Hz, 1H), 7.59-7.60 (d, J = 7.82 Hz, 1H), 7.72 (s, 1H), 7.84 dd, J = 8.31, 0.97 Hz, 2H), 8.32 (s, 1H); 13 C NMR (125 MHz; CDCl 3)? -0.00, 0.11, 0.26, 0.54, 30.63 (3), 34.61 (2), 49.19, 68.45, 77.09, 79.28, 87.24, 94.71, 99.46, 125.63, 131.74, 133.32 , 134.59, 135.23, 138.10, 141.41, 141.46, 144.66, 145.99, 153.87, 157.99, 159.53, 171.15; mp = 68-70 [deg.] C.
Step 5: ((2R, 3R, 4R, 5R) -3,4-Bis (tert-butyldimethylsilyloxy) -5- (6- (3- iodobenzylamino) -9H-purin- ) Tetrahydrofuran-2-yl) methanol (18)
The intermediate compound (1.28 g, 1.56 mmol) prepared in step 4 was dissolved in anhydrous methanol (100 mL), 25% sodium methoxide / methanol (15 mL) was added, and the mixture was stirred at room temperature for 12 hours. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure, and the obtained residue was separated by column chromatography to obtain the intermediate compound ((2R, 3R, 4R, 5R) -3,4- bis (tert- butyldimethylsilyloxy) Yl) tetrahydrofuran-2-yl) methanol (960 mg, 86.48%) was obtained as a pale-yellow amorphous solid.
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; CDCl 3 ) δ ppm -0.58 (s, 3H), -0.13 (s, 3H), 0.11-0.13 (d, J = 7.33 Hz, 6H), 0.74 (s, 9H), 0.95 (s, 9H), 3.68-3.71 (d, J = 12.71 Hz, 1H), 3.92-3.95 (d, J = 12.71 Hz, (D, J = 7.33, 4.40 Hz, 1H), 5.75-5.77 (d, J = 7.82 Hz, 1H), 6.39 (br s). J = 7.82 Hz, 1H), 7.30-7.32 (d, J = 7.82 Hz, 1H), 7.59-7.61 (d, J = 7.82 Hz, 1H), 7.70 (s, 1H), 7.76 (br s, 1H), 8.35 (br s, 1H); 13 C NMR (125 MHz; CDCl 3 ) δ 0.00, 1.30, 1.35, 1.38, 31.62 (3), 31.75 (3), 35.61 (2), 49.54, 68.95, 79.91, 79.96, 95.50, 96.96, 100.51, 127.37, 132.70, 136.30, 142.42, 142.57, 146.54, 146.61, 153.78, 158.46, 160.79; mp = 82-86 [deg.] C.
Step 6: (2S, 5R) -3,4-Bis (tert-butyldimethylsilyloxy) -5- (6- (3- iodobenzylamino) -9H- purin- Preparation of tetrahydrofuran-2-carboxamide (19)
The intermediate compound (450 mg, 0.63 mmol) prepared in Step 5 and pyridinium dichromate (5.47 g, 14.54 mmol) were dissolved in DMF (50 mL) under a nitrogen stream, followed by stirring at room temperature for 12 hours. After confirming completion of the reaction, the resulting solid was washed with water to obtain an acid intermediate. The obtained intermediate (450 mg, 0.62 mmol) was dissolved in anhydrous ethanol (10 mL) under a nitrogen stream, cooled to 0 ° C, thionyl chloride (0.25 mL, 3.10 mmol) was slowly added dropwise and the mixture was stirred at room temperature for 5 hours Lt; / RTI & gt; After completion of the reaction was confirmed, the reaction solution was concentrated under reduced pressure, and the residue was partitioned into ethyl acetate / water. The organic layer was washed with water and saturated aqueous sodium chloride solution, and dried over anhydrous magnesium sulfate. After concentration under reduced pressure, an intermediate ethyl ester was obtained. The ethyl ester thus obtained is added with a methylamine / 2N-THF solution under a nitrogen stream, followed by stirring at room temperature for 12 hours. After confirming the completion of the reaction, the reaction mixture was concentrated under reduced pressure and the residue was purified by column chromatography to obtain the intermediate compound (2S, 5R) -3,4-bis (tert-butyldimethylsilyloxy) -5- -9H-purin-9-yl) -N-methyltetrahydrofuran-2-carboxamide (400 mg, 85.65%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; CDCl 3 ) δ ppm -0.61 (s, 3H), -0.16 (s, 3H), 0.15 (s, 3H), 0.25 (s, 3H), 0.71 (s, 9H), 0.97 (s, 9H), 2.93-2.94 (d, J = 4.40 Hz, 3H), 4.33-4.34 (d, J = 3.42 Hz, (D, J = 7.33 Hz, 1H), 6.42 (br s, 1H), 7.03-7.06 (t, J = 7.82 Hz, 1H), 7.30-7.32 ), 7.59-7.61 (d, J = 7.82 Hz, 1H), 7.70 (s, 1H), 7.75 (s, 1H), 8.36 , 1H); 13 C NMR (125 MHz; CDCl 3 ) δ 0.00, 1.19, 1.21, 1.40, 23.71, 24.00, 31.53 (3), 31.58, 31.78 (2), 35.64, 49.60, 78.09, 81.25, 92.53, 95.48, 100.56, 127.30 , 132.72, 136.34, 142.44, 142.61, 146.64, 146.79, 154.27, 158.70, 160.96, 175.92; mp = 80-84 [deg.] C.
Step 7: (2S, 5R) -3,4-Dihydroxy-5- (6- (3-iodobenzylamino) -9H- purin-9- yl) -N- methyltetrahydrofuran- Manufacture of Radiate (20)
The intermediate compound (65 mg, 0.08 mmol) prepared in Step 6 was dissolved in anhydrous THF under a nitrogen stream, and then tetrabutylammonium fluoride (TBAF) (0.44 mL, 0.43 mmol, (1 M solution THF) Stir at room temperature for 1 hour. After confirming the completion of the reaction, the reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by column chromatography to obtain the intermediate compound (2S, 5R) -3,4-dihydroxy-5- (6- (3-iodobenzylamino) -9H-purin-9-yl) -N-methyltetrahydrofuran-2-carboxamide (52 mg, 95.45%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; DMSO-d 6 ) δ ppm 2.70-2.71 (d, J = 4.40 Hz, 3H), 4.14-4.16 (t, J = 3.91 Hz, 1H), 4.31 (s, 1H), 4.57 (D, J = 4.40 Hz, 1H), 4.67 (d, 1H), 5.96-5.97 (d, J = 7.33 Hz, 1H), 7.09-7.12 (t, J = 7.82 Hz, 1H), 7.35-7.36 (d, J = 7.82 Hz, 1H), 7.56-7.58 1H, J = 7.82 Hz, 1H), 7.72 (s, 1H), 8.29 (s, 1H), 8.42 (s, 1H), 8.53 (br s., 1H), 8.85-8.86 (m, 1H); 13 C NMR (125 MHz; DMSO-d 6 )? 25.81, 42.74, 72.56, 73.49, 85.09, 88.26, 95.06, 120.42, 127.11, 130.95, 135.86, 136.13, 141.17, 143.10, 148.70, 152.94, 154.86, 170.34; mp = 178-182 [deg.] C.
Step 8: (S) -3-Hydroxy-2 – ((R) -2-hydroxy-1- (6- (3-iodobenzylamino) -9H- purin- Preparation of N-methylpropanamide (12)
The intermediate compound (52 mg, 0.10 mmol) prepared in the above Step 7 was treated in the same manner as in Step 5 of Example 1 to obtain the title compound (35 mg, 83.33%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; DMSO-d 6 .) Δ ppm 2.35 (s, 3H), 3.5-3.6 (m, 1H), 3.72-3.74 (d, J = 9.29 Hz, 1H), 3.86 (br s. 2H), 4.99 (br s, 1H, exchangeable with D2O, OH), 5.21 (br. S., 1H), 4.00 (d, J = (d, J = 6.84 Hz, 1H), 7.56 d, J = 6.35 Hz, 2H, exchangeable with D 2 O, NH), 7.71 (s, 1H), 8.22 (s, 1H), 8.30 (s, 1H), 8.37 (br. s., 1H, exchangeable with D2O, NH); 13 C NMR (125 MHz; DMSO-d 6 ) δ 25.53, 42.81, 61.98, 62.26, 80.30, 84.62, 95.09, 119.43, 127.11, 130.91, 135.81, 136.16, 140.19, 143.31, 149.79, 152.98, 154.66, 169.42; HRMS (FAB) m / z calcd for C 18H 21 IN 6 O 4 [M + Na] +512.0669, found 535.0578; mp = 176-182 [deg.] C.
Production Example 4: Synthesis of Compound (21) ((R) -2- (2-hydroxy-1- (6- (3-iodobenzylamino) -9H- purin- 3-diol)
Step 1: ((3aR, 4R, 6R, 6aR) -6- (6- (3-Iodobenzylamino) -9H-purin-9- yl) -2,2-dimethyltetrahydrofuro [ 4-d] [1,3] dioxol-4-yl) methanol (22)
The intermediate compound (1.73 g, 2.75 mmol) prepared in the step 2 of Example 1 was treated in the same manner as in the step 5 of Example 3 with (3aR, 4R, 6R, 6aR) -6- L, 3-dioxol-4-yl) methanol (1.04 g, 93.69 & lt; RTI ID = 0.0 & gt; %).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; CDCl 3 ) δ ppm 1.36 (s, 3H), 1.63 (s, 3H), 3.75-3.80 (t, J = 11.24 Hz, 1H), 3.95-3.97 (d, J = 12.71 Hz , 5.19 (br, s, 2H), 5.10-5.11 (d, J = 4.89 Hz, 1H), 5.19 = 3.91 Hz, 1H), 6.58 (br s, 2H), 7.01-7.04 (t, J = 7.82 Hz, 1H), 7.29-7.30 (d, J = 6.84 Hz, 1H), 7.58-7.59 , J = 7.33 Hz, 1H), 7.69 (br s, 2H), 8.33 (br s, 1H); 13 C NMR (125 MHz; CDCl 3 ) δ 25.24, 27.66, 29.65, 63.36, 81.68, 83.03, 86.12, 94.26, 94.54, 113.93, 121.24, 126.80, 130.32, 136.52, 136.58, 139.71, 140.72, 147.66, 152.73, 154.94 ; mp = 72-76 [deg.] C.
Step 2: (2R, 3S, 4R, 5R) -2- (hydroxymethyl) -5- (6- (3-iodobenzylamino) -9H- purin-9- yl) tetrahydrofuran- – Preparation of diol (23)
The intermediate compound (250 mg, 0.47 mmol) prepared in the above step 1 was dissolved in 80% acetic acid (250 mL), and the mixture was heated under reflux at 100 ° C for 12 hours. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure, toluene (4 x 50 mL) was added, and the mixture was concentrated under reduced pressure. The obtained residue was purified by column chromatography to obtain the intermediate (2R, 3S, 4R, Methyl) -5- (6- (3-iodobenzylamino) -9H-purin-9-yl) tetrahydrofuran-3,4-diol (177 mg, 76.95%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; DMSO-d 6 )? Ppm 3.56 (br s, 1H), 3.67-3.69 (d, J = 10.27 Hz, 1H), 3.97 ), 4.61-4.67 (m, 3H), 5.17 (d, J = 2.93 Hz, 1H, exchangeable with D 2 O, OH), 5.35-5.36 (t, J = 5.38 Hz, 1H, exchangeable with D 2 O, OH), 5.43-5.44 (d, J = 5.38 Hz, 1H, exchangeable with d 2O, OH), 5.89-5.90 (d, J = 4.89 Hz, 1H), 7.09-7.11 (t, J = 7.33 Hz, 1H), 7.35-7.36 (d, J = 6.84 Hz, 1H), 7.56-7.58 (d, J = 7.33 Hz, 1H), 7.72 ), 8.45 (br s, 1H, exchangeable with D 2 O, NH); 13 C NMR (125 MHz; DMSO-d 6) [delta] 42.69, 62.08, 71.06, 73.97, 86.32, 88.42, 95.09, 120.24, 127.08, 130.91, 135.81, 136.16, 140.47, 143.22, 149.00, 152.76, 154.79; mp = 174-178 [deg.] C.
Step 3: (R) -2- (2-Hydroxy-1- (6- (3-iodobenzylamino) -9H-purin-9-yl) ethoxy) propane- )
The intermediate compound (77 mg, 0.15 mmol) prepared in the above Step 2 was treated in the same manner as in Step 5 of Example 1 to obtain the desired compound (62 mg, 80.51%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; CD 3 OD)? Ppm 3.42 – 3.44 (d, J = 5.38 Hz, 2H), 3.54-3.58 (m, 1H), 3.65-3.68 ), 3.75-3.78 (dd, J = 11.73,4.44 Hz, 1H), 4.01-4.02 (d, J = 5.38 Hz, 2H), 4.53 (s, 2H), 6.04-6.06 J = 7.82 Hz, 1H), 7.76 (s, 1H), 7.07-7.10 (t, J = 7.82 Hz, 1H), 7.38-7.40 (d, J = 7.82 Hz, 1H), 7.59-7.60 ), 8.27 (s, 1 H), 8.29 (s, 1 H); 13 C NMR (125 MHz; CD 3 OD)? 42.88, 60.80, 61.25, 62.76, 80.26, 84.02, 93.52, 119.00, 126.44, 129.93, 135.90, 136.10, 139.65, 141.72, 148.97, 152.52, 154.53; HRMS (FAB) m / z calcd for C 17 H 20 IN 5 O 4 [M + H] +485.0560, found 486.0625; mp = 72-76 [deg.] C.
PATENT
WO 2008111082
REFERENCES
1: Avni I, Garzozi HJ, Barequet IS, Segev F, Varssano D, Sartani G, Chetrit N, Bakshi E, Zadok D, Tomkins O, Litvin G, Jacobson KA, Fishman S, Harpaz Z, Farbstein M, Yehuda SB, Silverman MH, Kerns WD, Bristol DR, Cohn I, Fishman P. Treatment of Dry Eye Syndrome with Orally Administered CF101 Data from a Phase 2 Clinical Trial. Ophthalmology. 2010 Mar 19. [Epub ahead of print] PubMed PMID: 20304499.
2: Bar-Yehuda S, Rath-Wolfson L, Del Valle L, Ochaion A, Cohen S, Patoka R, Zozulya G, Barer F, Atar E, Piña-Oviedo S, Perez-Liz G, Castel D, Fishman P. Induction of an antiinflammatory effect and prevention of cartilage damage in rat knee osteoarthritis by CF101 treatment. Arthritis Rheum. 2009 Oct;60(10):3061-71. PubMed PMID: 19790055.
3: Borea PA, Gessi S, Bar-Yehuda S, Fishman P. A3 adenosine receptor: pharmacology and role in disease. Handb Exp Pharmacol. 2009;(193):297-327. Review. PubMed PMID: 19639286.
4: Moral MA, Tomillero A. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2008 Mar;30(2):149-71. PubMed PMID: 18560631.
5: Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, Molad Y, Rosner I, Rozenbaum M, Mader R, Adawi M, Caspi D, Tishler M, Langevitz P, Rubinow A, Friedman J, Green L, Tanay A, Ochaion A, Cohen S, Kerns WD, Cohn I, Fishman-Furman S, Farbstein M, Yehuda SB, Fishman P. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: data from a phase II clinical trial. J Rheumatol. 2008 Jan;35(1):41-8. Epub 2007 Nov 15. PubMed PMID: 18050382
/////////////CF 101, Piclidenoson, CF101, CF-101, CF 101, ALB-7208, ALB 7208, ALB7208, IB MECA, Phase III, Plaque psoriasis, Rheumatoid arthritis, UNII-30679UMI0N, Пиклиденозон , بيكليدينوسون , 匹利诺生 , Can-Fite BioPharma
CNC(=O)[C@H]1O[C@H]([C@H](O)[C@@H]1O)N1C=NC2=C(NCC3=CC(I)=CC=C3)N=CN=C12














