














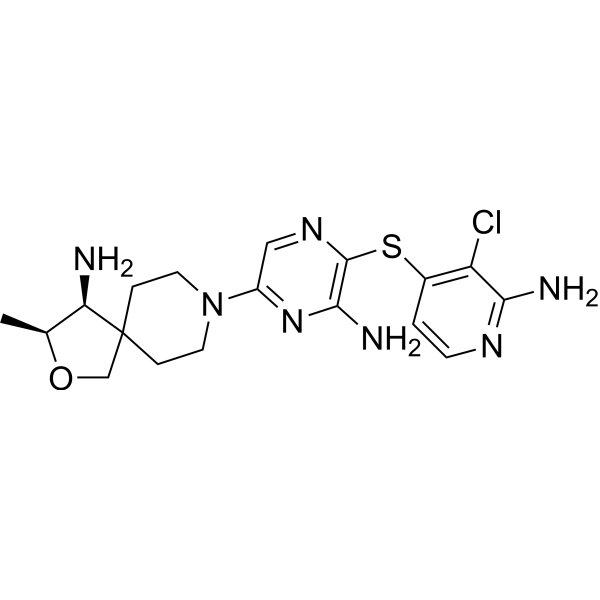
TNO 155
2-Oxa-8-azaspiro[4.5]decan-4-amine, 8-[6-amino-5-[(2-amino-3-chloro-4-pyridinyl)thio]-2-pyrazinyl]-3-methyl-, (3S,4S)-
- (3S,4S)-8-[6-Amino-5-[(2-amino-3-chloro-4-pyridinyl)thio]-2-pyrazinyl]-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine
- (3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine
| Molecular Weight |
421.95 |
|---|---|
| Formula |
C₁₈H₂₄ClN₇OS |
| CAS No. |
- PTPN11 inhibitor TNO155
- SHP2 inhibitor TNO155
- TNO-155
- TNO155
- UNII-FPJWORQEGI
TNO155 is a potent selective and orally active allosteric inhibitor of wild-type SHP2 (IC50=0.011 µM). TNO155 has the potential for the study of RTK-dependent malignancies, especially advanced solid tumors.
- Originator Novartis
- Developer Mirati Therapeutics; Novartis
- Class Antineoplastics
- Mechanism of ActionProtein tyrosine phosphatase non receptor antagonists
- Phase I/IISolid tumours
- Phase IColorectal cancer
- 11 Jul 2021Phase I trial in Solid tumours is still ongoing in USA, Canada, Japan, South Korea, Netherlands, Singapore, Spain, Taiwan (NCT03114319)
- 04 Jun 2021Efficacy, safety and pharmacokinetics data from phase I trial in Solid tumours presented at 57th Annual Meeting of the American Society of Clinical Oncology (ASCO-2021)
- 08 Jan 2021Novartis plans a phase Ib/II trial for Solid tumours (Combination therapy, Inoperable/Unresectable, Late-stage disease, Metastatic disease, Second-line therapy or greater) in February 2021 (NCT04699188)
CLIP
Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling
Results: In EGFR-mutant lung cancer models, combination benefit of TNO155 and the EGFRi nazartinib was observed, coincident with sustained ERK inhibition. In BRAFV600E colorectal cancer models, TNO155 synergized with BRAF plus MEK inhibitors by blocking ERK feedback activation by different RTKs. In KRASG12C cancer cells, TNO155 effectively blocked the feedback activation of wild-type KRAS or other RAS isoforms induced by KRASG12Ci and greatly enhanced efficacy. In addition, TNO155 and the CDK4/6 inhibitor ribociclib showed combination benefit in a large panel of lung and colorectal cancer patient–derived xenografts, including those with KRAS mutations. Finally, TNO155 effectively inhibited RAS activation by colony-stimulating factor 1 receptor, which is critical for the maturation of immunosuppressive tumor-associated macrophages, and showed combination activity with anti–PD-1 antibody.
Conclusions: Our findings suggest TNO155 is an effective agent for blocking both tumor-promoting and immune-suppressive RTK signaling in RTK- and MAPK-driven cancers and their tumor microenvironment. Our data provide the rationale for evaluating these combinations clinically.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
PATENT
WO 2015107495
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015107495
PATENT
WO 2020065453
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020065453
(3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine, which has the formula I,
WO/2015/107495 A1 describes a method for the manufacture of the compound of the formula I which can be characterized by the following reaction scheme 1:
[0008] The last compound resulting from step g above was then reacted as in the following scheme 2:
Scheme 2:
[0009] Thus the compound of formula I is obtained (last compound in the scheme 2, above). The synthesis requires at least the 9 steps shown and is rather appropriate for synthesis in laboratory amounts.
Scheme 1A:
[0016] Therefore, the process, though readily feasible on a laboratory scale, is not ideal for manufacture at a large scale.
[0017] The compound added in reaction b in Scheme 2 is obtained in WO
2015/107495 A1 as “Intermediate 10” follows:
Scheme 3:
[0018] An issue here is the relatively low yield of the amine resulting from reaction a in
Scheme 3.
[0019] In addition, while WO 2015/107495 A1 generically mentions that pharmaceutically acceptable salts of the compound of the formula I may be obtainable, no concrete reason for obtaining such salts and no specific examples of salts are described.
[0020] In addition, given the many potentially salt forming groups in formula I, it is not clear whether any salts with a clear stoichiometry can be formed at all.
Example 1
Method of synthesis of the compound of the formula I ((3S,4S)-8-(6-amino-5-((2-amino-3- chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine):
The overall synthesis can be described by the following Reaction Scheme A:
Scheme A:
Step a
[00293] To a solution of A1 (10.4 kg, 100 mol, 1.0 Eq) in CH2Cl2 (50 L) was added imidazole (8.16 kg, 120 mol, 1.2eq) and TBSCl (18 kg, 120 mol, 1.2 Eq) at 0 °C. After addition, the mixture was stirred at 0°C for 4 h . GC showed the reaction was finished. (A1/ (A1 + A2) < 1%). The reaction mixture was quenched with saturated NaHCO3 (14L) at 0-5°C. Phases were separated. The organic phase was washed with brine (14L). The organic layer was dried over Na2SO4, concentrated under vacuum at 40-45°C to afford A2 (23.3 kg, assay 88%, yield 94%) which was used for the next step directly. 1H NMR (400 MHz, CDC13) δ = 4.35 (d, J= 8.8 Hz, 1H), 3.74 (s, 3H), 2.48 (s, J= 8.8
Hz, 3H), 0.93 (s, 9H), 0.09 (s, 6H).
Step b
[00294] To a solution of A2 (7.5 kg, 34.3 mol, 1.0 Eq) and N,O-dimethylhydroxylamine hydrochloride (6.69 kg, 68.6mol, 2.0 Eq) in THF (20 L) was added drop-wise a solution
of chloro(isopropyl)magnesium (2 M, 51.45 L, 3.5 Eq) at 0 °C under N2 over 5-6 h. After addition, the reaction mixture was stirred at 0 °C for 1h, GC showed the reaction was finished (A2/(A2+A3) < 2 %). The mixture was quenched with NH4Cl (25 L) slowly by keeping the temperature at 0-5°C. After addition, the reaction mixture was stirred for 30min. Phase was separated. The aqueous layer was extracted with EA(2 x 20 L). The combined organic phase was washed with brine (25L), dried over Na2SO4, concentrated to give A3(9.4 kg, assay 86%, yield 95%) which was used for the next step directly. 1HNMR (400 MHz, CDCl3) δ = 4.67 (m, J= 6.6 Hz, 1H), 3.70 (s, 3H), 3.21 (s, 3H), 3.17 (d, 3H)2.48 (s , J= 6.6 Hz, 3H), 0.90 (s, 9H), 0.10 (s, 3H), 0.08 (s, 3H).
[00295] To a solution of A3 (7.1 kg, assay 86%, 24.65 mol, 1.0 Eq) in DCM (30 L) was added dropwise a solution of LiAlH4 (2.4 M, 11.3 L, 1.1 Eq) at -70 °C under N2. Then the reaction mixture was stirred at -70 °C for 3h, and TLC showed the reaction was finished (PSC-1). The mixture was warmed to 0 °C, and then quenched with sat. potassium sodium tartrate (35 L) at 0 °C. After addition, DCM (20L) was added and stirred for 2h at 20-25°C. Phases were separated. The aqueous layer was extracted with DCM (25 L). The combined organic phase was charged with sat. citric acid (45L) and stirred at 0°C for 8h. Phase was separated. The organic phase was washed with NaHCO3 (25L), brine (25 L), dried over Na2SO4, and the solvent was removed under vacuum at 25-30°C. n-Heptane (10 L) was added to the residue and concentrated under vacuum at 30-35°C. n-Heptane (10 L) was added to the residue again and concentrated under vacuum at 30-35°C to give A4 (4.2 kg, assay
60%, yield 54%) which was used for the next step directly.
Step d
[00296] To a solution of diisopropylamine (3.06 kg, 30.3 mol, 1.5 eq) in THF (20 L) cooled to approximately -10°C was added 2.5 M n-BuLi (12.12 L, 30.3 mol, 1.5 eq) under N2. The resulting mixture was stirred at approximately -10 °C for 30min, then a solution of A5 (5.2 kg, 20.20 mol, 1.0eq) in THF (10 L) was added slowly. After addition, the reaction mixture was stirred at -10°C for 30 min, and then cooled to -50°C. A4 (4.18 kg, 22.22 mol, 1.1eq) was added dropwise. After addition, the reaction mixture was stirred at -50°C for 30 min. The mixture was quenched with saturated aqueous NH4Cl (30L) and water (10L) at -50°C. The reaction mixture was warmed to 20-25°C. Phase was separated. The aqueous phase was extracted with EA (3 x 20 L). All organic phases were combined and washed with brine(20L), then concentrated to a yellow oil which was purified by column (silica gel, 100-200 mesh, eluted with n-heptane:EA from 50:1 to 10:1) to give A6 (5.5 kg, assay 90 %, yield 55%) as pale yellow oil. 1H NMR (400 MHz, CDCl3) δ = 4.35-4.15 (m, 2H), 3.95-3.74 (m, 3H), 3.52 (m, 2H), 2.67(m, 2H), 2.12-1.98 (m, 2H), 1.75-1.52 (m, 4H), 1.49 (s, 9H), 1.35-1.10 (m, 6H), 0.98 (s,
9H), 0.02 (s, 6H).
Step e
[00297] To a solution of A6 (11.4 kg, 25.58 mol, 1.0eq) in THF (60 L) was added LiBH4
(836 g, 38.37 mol, 1.5eq) in portions at 5-10 °C, and the reaction mixture was stirred at 20-25 °C for 18 h. HPLC showed the reaction was finished (A6/(A6+A7)<2%). The mixture was cooled to l0°C and slowly quenched with saturated NaHCO3 solution (15 L) and water (25L) with vigorously stirring. After gas formation stopped, vacuum filtration was applied to remove solids. The solid was washed with EA (2 x 15 L). Phase was separated; the aqueous phase was extracted with EA (3 x15L). All organic phases were combined and washed with brine (15L), and concentrated to obtain crude A7 (13.8 kg, assay 58%, yield 77%) which was used for the next step directly.
[00298] To a solution of A7 (8 kg, 19.82 mol, 1.0 eq) in THF (40 L) under nitrogen atmosphere was added TsCl (5.28 kg, 27.75 mol, 1.4 eq) at 10-15°C. After addition, the mixture was cooled to 0 °C, and 1M LiHMDS (29.7 L, 29.73 mol, 1.5 eq) was added dropwise during 2h. After addition, the mixture was stirred at 0°C for 3h. HPLC showed the reaction was finished (PSC-1 A7/ (A7+A8)<7%). TBAF (20.72 kg, 65.67 mol, 3.3 eq) was added into the mixture at 0 °C and the reaction mixture was stirred at 25-30 °C for 48h. HPLC showed the reaction was finished ( PSC-2, A9-intermedaite/(A9-intermediate+A9) < 2%). The mixture was quenched with saturated aqueous sodium bicarbonate solution (32L) and stirred for 30min at 0 °C. Phase was separated, and the aqueous phase was extracted with EA (3 x 20 L). The combined organic phase was washed with brine(20 L), dried over Na2SO4, and concentrated to a yellow oil which was purified by column (eluted with n-heptane:EA from 10:1 to 1:1) to give A9 (4.42 kg, assay 90%, yield 74 %) as pale yellow solid.
Step g
[00299] To a solution of A9 (4.0 kg, 14.74 mol, 1.0 eq) in DCM (40 L) cooled on an ice-bath was added DMP (9.36 kg, 23.58mol, 1.6eq) in portions, and it resulted in a suspension. After addition, the mixture stirred for 4 hours at 20-25°C. HPLC showed the reaction was finished (A9/(A9+A10)<2%). DCM (30L) was added at 0°C. After addition, the mixture was quenched with saturated aqueous Na2SO3 (20 L). The mixture was stirred for 30min at 0 °C, filtered and the white solid was washed with DCM (2 x15L). Phase was separated, and the organic phase was cooled to 0°C, to which was added saturated aqueous NaHCO3 (20L) and stirred for 1h. Phase was separated, and the organic phase was washed with brine(25L), dried over Na2SO4, and concentrated to a yellow oil which was purified by column (eluted with n-heptane:EA from 50:1 to 10:1) to give A10 (3.70 kg, assay 88%, ee value 95.3%, yield 82%) as white solid. 1H NMR (400 MHz, DMSO-d6) δ = 4.20 (d, J = 8.0 Hz,
1H), 3.98-3.67 (m, 4H), 3.08-2.90 (m, 2H), 1.54-1.39(m, 13H), 1.18 (d, J = 8.0 Hz, 3H).
Step h
[00300] To a solution of A10 (4.60 kg, 17.08 mol, 1.0 eq) in THF (40 L) was added
Ti(OEt)4 (15.58 kg, 68.32 mol, 4.0 eq) and (R)-t-Butyl sulfmamide (4.14 kg, 34.16 mol, 2.0 eq) at 25 °C. After addition, the mixture was heated to 70°C and stirred for 20h. HPLC showed the reaction was finished (PSC-l, A10/(A10+A12)<4%). The mixture was cooled to -30— 40°C, and MeOH (4 L) was added dropwise within 30 min and stirred for 1 h. 2M L1BH4 (8.1 L) solution was added dropwise to the reaction mixture at -40- -50°C and stirred for 1h. HPLC indicated all of imine was consumed (PSC-2, A12/(A12+A13)<1%). The mixture was warmed to -30 °C and stirred for 1h, then warmed to 0 °C within 2 h and stirred for 1h, then warmed to 20-25 °C and stirred for 30min. IP AC ( 25L) was added to above mixture, NaHCO3(5L) was added dropwise in about 1h at 25 °C and stirred for 30 min. The mixture was filtered under vacuum and the cake was washed with IP AC (8 x15L). The combined organic phase was washed with brine (25L), then evaporated under vacuum to get a solution of A13
(about 28kg) which was used for next step.
[00301] To a mixture of A13 in IPAC (about 28 kg, 17.08 mol, 1.0 eq) was added dropwise
4M HCl/IPA (8.54 L, 34.16 mol, 2.0 eq) at -5 °C and stirred for 5h at -5 °C. HPLC showed that A13 was consumed completely (A13/(A14+A13)<1%). MTBE (25 L) was added to above mixture within
30 min and stirred for 30 min at -5 °C .The solid was collected by vacuum filtration. The cake was washed with MTBE (2 x 2.5 L). The wet cake was used for next step directly.
Step j
[00302] The wet solid A14 (from 9.2 kg A10) was stirred in MTBE(76 L) at 25°C, then the
16% NaOH (9.84 kg) solution was added dropwise to the MTBE suspension while maintaining IT<10ºC. After addition, the mixture was stirred for 15 min and all solids were dissolved at 0°C. The organic phase was separated, and the aqueous phase was extracted with MTBE (2 x 20L). The combined organic phase was washed with brine (10 L) and evaporated under vacuum to remove all MTBE. ACN (24 L) was added to above residue, and the mixture was evaporated under vacuum to remove the organic solvents and yielded a crude A15 (5.42 kg, qnmr 90%, 18.04 mol, 1.0 eq). ACN (34.68 kg) was added to above residue and stirred for 10 min at 65°C. A solution of (-)-O-acetyl-D-mandelic acid (3.15kg,16.2 mol, 0.9 eq) in ACN(11.6 kg) was added drop-wise to the mixture (firstly added 1/3, stirred for 0.5 h, then added the others) over 3h. The mixture was stirred for 1 h at 65°C, then cooled to 25°C over 4h and stirred for l2h at 25°C . The solid was collected by vacuum filtration, and the cake was washed with pre-cooled ACN (2 x15kg) (PSC-1) and dried under vacuum to give
A16 (7.36 kg, yield 46% from A10 to A16). 1H NMR (400 MHz, DMSO-d6) δ = 7.43-7.29 (m, 5H),
5.58 (s, 2H), 4.12-4.07 (m, 1H), 3.75-3.65 (m, 3H), 3.51-3.49 (m, 1H), 3.18-3.17 (m, 1H), 2.84 (bs,
2H), 2.05 (s, 3H), 1.60-1.40 (m, 13H), 1.14-1.12 (d, J= 8.0 Hz, 3H).
Step k
[00303] To a solution of A16 (15 g) in MeOH (90 mL) was added dropwise 5N HC1/IPA
(45 mL) at room temperature within 15 minutes. After the addition, the mixture was stirred for 6 hours.
IP AC (180 mL) was added dropwise to above mixture within 1h at room temperature. The resulting mixture was stirred for another 30 minutes before it was cooled to 0-5 °C. The mixture was stirred at 0- 5 °C for another 2h and the precipitants were collected by filtration. The cake was washed with (45*2 mL) IP AC, dried under vacuum at 60 °C overnight to afford the product as a white solid. 1H NMR (400
MHz, DMSO-d6) δ = 9.37 (br s, 1H), 9.25 (br s, 1H), 8.42 (br s, 3H), 4.26 – 4.17 (m, 1H), 3.72 (ABq, J
= 9.1 Hz, 2H), 3.50 – 3.41 (m, 1H), 3.28 – 3.18 (m, 1H), 3.18 – 3.09 (m, 1H), 2.99 – 2.74 (m, 2H), 2.07 – 1.63 (m, 4H), 1.22 (d, J= 6.5 Hz, 3H).
Step l
[00304] To a mixture of A17 (10 g) and Z17a (9.5 g) in DMAC (60 mL) was added K2CO3
(22.5 g) and H2O (40 mL) at room temperature. The mixture was degassed with nitrogen and stirred at
90 °C overnight. The mixture was cooled to room temperature, diluted with Me-THF (500 mL) and
H2O (280 mL). The organic phase was separated and the aqueous phase was extracted with Me-THF
(300 mL*2). The combined organic phases were washed with brine (200 mL*3), concentrated under
vacuum to remove most of the solvent. The residue was diluted with IPA (60 mL) and H2O (20 mL), stirred at 50 °C for 1h, cooled to 5 °C within 3h, stirred at this temperature for 1h. The solid was collected by vacuum filtration, dried under vacuum to afford the product as a yellow solid (l2g,
87.4%). 1H NMR (400 MHz, DMSO-d6)δ = 7.64 (d, J= 6.2 Hz, 1H), 7.62 (s, 1H), 6.26 (s, 2H), 6.13 (s, 2H), 5.74 (d, J= 5.3 Hz, 1H), 4.12 – 4.02 (m, 1H), 3.90 – 3.78 (m, 2H), 3.67 (d, J= 8.4 Hz, 1H), 3.49 (d, J= 8.4 Hz, 1H), 3.33 (s, 2H), 2.91 (d, J= 5.1 Hz, 1H), 1.78 – 1.68 (m, 1H), 1.67 – 1.57 (m, 1H), 1.56 – 1.41 (m, 2H), 1.08 (d, J= 6.5 Hz, 3H).
Example 2
Formation of the succinate salt of the compound of the formula I:
[00305] The reaction is summarized by the following Reaction Scheme:
[00306] To a mixture of A18 (10 g) in MeOH (76 g) and H2O (24 g) was added succinic acid (2.94 g) at room temperature. The mixture was heated to 50 °C and stirred for 30 minutes to dissolve all solid. The solution was added to IPA (190 mL) at 60-65 °C. The resulting mixture was stirred at 60 °C >5 hours, cooled to -15 °C within 5 hours and stirred at this temperature >4 hours. The solid was collected by vacuum filtration, dried under vacuum to afford the product as an off-white solid(l0.8 g, 82.8%). 1H NMR (400 MHz, DMSO-d6)δ = 7.64 (d, J= 6.2 Hz, 1H), 7.63 (s, 1H), 6.26 (s, 2H), 6.16 (s, 2H), 5.74 (d, J= 5.3 Hz, 1H), 4.12 – 4.02 (m, 1H), 3.90 – 3.78 (m, 2H), 3.67 (d, J= 8.4 Hz, 1H), 3.49 (d, J= 8.4 Hz, 1H), 3.33 (s, 2H), 2.91 (d, J= 5.1 Hz, 1H), 2.34 (s, 4H), 1.71 – 1.60 (m, 4H), 1.13 (d, J = 6.5 Hz, 3H).
[00307] In a special variant, the reaction follows the following Reaction Scheme, also including an optional milling to yield the final product:
Example 3
Formation of the intermediate Z17a (3-((2-amino-3-chloropyridin-4-yl)thio)-6-chloropyrazin-2- amine). Variant 1:
[00308] The compound Z17a was obtained by reaction according to the following Reaction
Scheme:
[00309] In detail, the synthesis of Compound Z17a was carried out as follows:
Step a
[00310] Under nitrogen atmosphere, n-BuLi (2.5M, 7.6 L) was added dropwise to a solution of 3-chloro-2-fluoropyridine (2 kg) in THF (15 L) at -78°C. Then the resultant mixture was stirred for 1h. Then a solution of I2 (4.82 kg) in THF (6 L) was added dropwise. After addition, the reaction mixture was stirred for 30 min, and then quenched with sat. Na2SO3 (10 L), and warmed to 20- 25°C. Phase was separated. The aqueous phase was extracted with EA (2 x 10 L). The combined organic phase was washed with sat.Na2SO3 (2 x 8 L), brine (8 L), and dried over Na2SO4. The organic phase was concentrated under vacuum. The residue was slurried in MeOH (4 L), filtered, and dried to offer 3-chloro-2-fluoro-4-iodopyridine 1c (2.2 kg, yield 68%).
Step b
[00311] Into a solution of Compound 1c (8 kg) in DMSO (48 L) was passed through NH3
(gas) at 80 °C overnight. TLC showed the reaction was finished. The reaction mixture was cooled to RT. The reaction mixture was added to water (140 L). The solid was collected and washed with water (25 L), dried to afford Z17b (6.91 kg, yield 87%). 1H NMR (400 MHz, CDC13) δ = 7.61 (d, J= 6.8 Hz,
1H), 7.14 (s , J= 6.8 Hz, 1H), 5.09 (bs, 2H).
Step c
[00312] A solution of 2-amino-6-chloro-pyrazine la (1 kg, 7.69 mol) in DCM (15 L) was heated to reflux, to which was charged NBS (4l7g) in portions during 1 h. The reaction was cooled to room temperature. The reaction mixture was washed with water (3 L) and brine (3 L). The organic phase was evaporated, and the residue was purified by column chromatography to give product Z17f
(3-bromo-6-chloropyrazin-2-amine) (180 g, 11% yield).
[00313] To a solution of 3-bromo-6-chloropyrazin-2-amine Z17f (6.0 kg, 28.78 mol) in 1,4- Dioxane (40 L) was added Pd(OAc)2 (64.56 g, 287.6 mmol), Xantphos (333 g, 575.6 mmol), and DIPEA (7.44 kg, 57.56 mol) at room temperature under nitrogen. After another 30 minutes purging with nitrogen, methyl 3-mercaptopropanoate (3.81 kg, 31.70 mol) was added, resulting in darkening of the orange mixture. The mixture was heated to 90°C. HPLC showed complete conversion of the starting material. The mixture was allowed to cool to about room temperature, then diluted with EtOAc (40L). After aging for 30 min with stirring, the entire mixture was filtered and solids were washed with EtOAc (3 x 15L). The combined orange filtrate was concentrated to dryness and the solid residue was suspended in DCM (45 L). The mixture was heated to 35-40 °C and stirred for 1h until all solids were dissolved. Then n-heptane (45L) was added dropwise. Upon complete addition, the mixture was cooled to 15-20 °C with stirring for 1h. The solids were collected by vacuum filtration and solids were washed with cold 1:1 DCM/heptane (25 L), then heptane (25 L) (PSC-2). The solids were dried over the weekend to give Z17d (5.32 kg, yield 75%). 1H NMR (400 MHz, CDCl3) δ = 7.83 (s, 1H), 4.88 (bs,
2H), 3.73 (s, 3H), 3.47 (t, J= 9.2 Hz, 2H), 2.79 (t, J= 9.2 Hz, 2H).
Step e
[00314] To a solution of Z17d (8.0 kg, assay 95%, 30.68 mol) in THF (70 L) was added
EtONa (prepared from 776 g Na and 13.6 L EtOH) at room temperature and the mixture was stirred at
ambient temperature for 1 hour. The mixture was then concentrated to a wet yellow solid by rotary evaporation and the residue was suspended in DCM (40L). The mixture stirred under N2 for l6h. The solids were collected by vacuum filtration and the cake was washed with DCM (about 15 L) until the filtrate was colorless (PSC-2). The solids were then dried under vacuum to give Z17c (6.93 kg, qNMR
72 %, yield 88%). 1H NMR (400 MHz, D2O) δ = 7.37 (s, 1H).
Step f
[00315] To a mixture of Z17c (6.95 kg, assay 72%, 27.23 mol) in l,4-dioxane (72 L) was added Xantphos (233 g, 411 mmol, 0.015 eq), Pd2(dba)3 (186 g, 206 mmol, 0.0075 eq), Z17b (7.13 kg, 28.02 mol) and DIPEA (7.02 kg, 54.46 mol). The system was vacuated and purged with nitrogen gas three times. The mixture was stirred at 65 °C for 16 h under N2. The mixture was cooled to RT and water (50 L) was added, filtered. The cake was washed with EA (25 L). The filtrate was extracted with EA (4 x 20 L). The organic phase was concentrated in vacuum to offer the crude product which was combined with the cake. Then DCM (60 L) was added to the crude product and stirred at 25-30°C for l8h and then filtered. The filter cake was slurried with CH2Cl2 (30 L) for 4 hrs and filtered. The filter cake was slurred in CH2Cl2 (30 L) for 16 hrs and filtered. Then the filter cake was dried in vacuum to give Z17a (3-((2-amino-3-chloropyridin-4-yl)thio)-6-chloropyrazin-2-amine; 9.1 kg, 84 %) as light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ = 7.89 (s, 1H), 7.7 (d, J= 7.6 Hz, 1H), 7.18 (bs, 2H), 6.40 (bs, 2H), 5.97 (d, J= 7.6 Hz, 1H).
Example 4
Alternative formation of the intermediate Z17a (here also named Y7a)
[00316] By way of alternative and according to a preferred reaction method, the compound of the formula Z17a was obtained according to the following Reaction Scheme:
In detail, the synthesis of the compound of the formula Y7a = Z17a was carried out as follows:
Step a
[00317] 2, 3, 5-trichloropyrazine (70.50 g, 384.36 mmol, 1 equiv) and ammonia solution
(25% wt, 364.00 g, 400 mL, 2.68 mol, 6.14 equiv) were added to a 1-L sealed reactor. The mixture was heated to 80 °C and stirred for 24 h, and the reaction was completed. The reaction mixture was cooled to 30 °C and filtered to give a brown filter cake. The brown filter cake was dissolved in acetone
(50 mL), and filtered. To the filtrate was added petroleum ether (300 mL). The suspension was stirred for 4 h, and filtered to give the crude product. The crude product was slurried in combined solvents of petroleum ether and acetone (10/1, 200 mL) and filtered to give the product Y7d (51.00 g, 307.91 mmol, 80% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ = 7.63 (s, 1H).
[00318] To a 200 mL round bottom flask was added Na2S (10.816 g, 44wt% containing crystalline water, 60.978mmol) and toluene (100 mL). The mixture was heated to reflux, and water was removed with a Dean-Stark trap (about 5~6 mL water was distilled out). After cooling, the mixture was concentrated to dryness.
[00319] To above round bottom flask was added Y7d (5.000 g, 30.489mmol) and 2-methylbutan-2-ol (50 mL), the reaction was heated to reflux and stirred for 36 h. After cooling to 25 °C, the mixture was filtered. The solvent of the filtrate was exchanged with n-heptane (5 V, 3 times, based on Y7d), and finally concentrated to IV residue. THF (25 mL) was charged to the residue at 25 °C and stirred. The suspension was filtered and washed with THF/n-heptane (5 mL/5 mL) to give a brown solid (6.200 g).
[00320] To another 200 mL round bottom flask was added above brown solid (6.200 g),
10% brine (25 mL), Me-THF (30 mL) and n-Bu4NBr (9.829 g, 30.489 mmol). The mixture was stirred for 0.5 h at room temperature, and the phases were separated. The organic phase was washed with 20% brine (25 mL), and exchanged the solvent with iso-propanol (5 V *3 times, based on Y7d) to give the iso-propanol solution of Y7c (27.000g, 99.2% purity by HPLC area, 58.08% assay yield). 1H NMR (400 MHz, DMSO-d6) δ = 6.88 (s, 1H), 2.97 – 2.92 (m, 14H), 1.38 – 1.31 (m, 14H), 1.13 – 1.04 (m,
14H), 0.73 – 0.69 (t, 21H).
Step c
[00321] To a 25-mL round-bottom flask was added Y7c (4.7g, 23.27wt%, IPA solution from Step b, 2.723 mmol, 1.0 equiv), Y7b (1.052 g, 4.085 mmol, 1.5 equiv), l,lO-Phenanthroline (0.05 g, 0.272 mmol) and water (8 mL). The mixture was purged with nitrogen gas three times, and Cul (0.026 g, 0.136 mmol) was added under nitrogen atmosphere. The mixture was heated up to 65 °C and stirred for 3 h, and the reaction was completed. The reaction was cooled to room temperature and filtered, and the filter cake was washed with water (4 mL*3). The filter cake was slurried in MTBE (6 mL) for 30 min and filtered. The filter cake was washed with MTBE (6 mL) and dried to afford Y7a which is Z17a (565 mg, 72% yield).
[00322] Z17b is synthesized as described in Example 3 Step a and Step b.
Example 5
Alternative Synthesis of the intermediate Z17a:
[00323] According to another preferred method, the compound of the formula Z17a was obtained in accordance with the following Reaction Scheme:
[00324] The reactions were carried out as follows:
Step a
Y7d was synthesised as described in Example 4 step a.
[00325] To a three-necked round-bottle flask was added Y7d (200 mg, 1.22 mmol, 1 equiv), dioxane (4 mL). The solution was vacuated and purged with nitrogen gas three times. Xantphos (14mg, 0.024 mmol, 0.02 equiv), PdCl2(dppf) (8.9 mg, 0.012 mmol, 0.1 equiv), and DIPEA (0.32 g, 2.44 mmol, 2.0 equiv) were added under nitrogen atmosphere. The solution was heated to 85 °C for overnight. The reaction was cooled and evaporated. The residue was purified by column chromatography (eluent/ethyl acetate/heptane = 1/1) to give Z17d (259 mg, 0.99 mmol, 81%). 1H NMR (400 MHz, CDCl3) δ = 7.83 (s, 1H), 4.88 (bs, 2H), 3.73 (s, 3H), 3.47 (t, J= 9.2 Hz, 2H), 2.79 (t, J= 9.2 Hz, 2H).
[00326] The remaining steps were carried out as described in Example 4, Steps e and f, to yield Z17a. Z17b was synthesized as described in Example 3 Step a and Step b.
Example 6
(3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8- azaspiro[4.5]decan-4-amine. succinate (1:1) hemihydrate. modification (form) HA:Variant a)
[00327] 50 ml ethanol and 2.5 ml water were added to a 100ml flask containing 3.0 g of free base of 3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (obtained as A18 for example as described in Example 1) and 848.0 mg of succinic acid. The mixture was heated to 50°C to generate a clear solution. The temperature was lowered to 15°C during a period of 3 hours. The solution was kept stirring at 15°C overnight.
Precipitated solid was separated via suction filtration and 50 ml of acetone was added to produce a suspension. The suspension was stirred at 50°C for 3 hours. The solid was separated with suction filtration and dried at room temperature under vacuum for 3 hours. Yield was about 60%.
[00328] The succinate appeared as a highly crystalline solid, with a melting point onset of
94.4°C and an accompanying enthalpy of 96 J/g. The succinate salt crystals showed aggregates of broken drusy tabular particles.
[00329] Variant b)
[00330] 14.34 g of 3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)- 3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine free form (obtained as A18 for example as described in Example 1) and 4.053 g of succinic acid were equilibrated in 100 mL 95% EtOH at 50°C. Add 5 mL of water into the system and heat to 70-75 °C. Add 95 mL of pure EtOH and heat for 30 min more. Stir over night at 25 oC. Filter the mixture wash with EtOH and dry under vacuum in an oven at room temperature. Yield is 87.5%.
PATENT
WO 2020065452
PATENT
PHARMACEUTICAL COMBINATION COMPRISING TNO155 AND NAZARTINIB
PAPER
Journal of Medicinal Chemistry (2020), 63(22), 13578-13594.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01170
SHP2 is a nonreceptor protein tyrosine phosphatase encoded by the PTPN11 gene and is involved in cell growth and differentiation via the MAPK signaling pathway. SHP2 also plays an important role in the programed cell death pathway (PD-1/PD-L1). As an oncoprotein as well as a potential immunomodulator, controlling SHP2 activity is of high therapeutic interest. As part of our comprehensive program targeting SHP2, we identified multiple allosteric binding modes of inhibition and optimized numerous chemical scaffolds in parallel. In this drug annotation report, we detail the identification and optimization of the pyrazine class of allosteric SHP2 inhibitors. Structure and property based drug design enabled the identification of protein–ligand interactions, potent cellular inhibition, control of physicochemical, pharmaceutical and selectivity properties, and potent in vivo antitumor activity. These studies culminated in the discovery of TNO155, (3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (1), a highly potent, selective, orally efficacious, and first-in-class SHP2 inhibitor currently in clinical trials for cancer.

file:///C:/Users/Inspiron/Downloads/jm0c01170_si_001.pdf
(3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8- azaspiro[4.5]decan-4-amine (1):
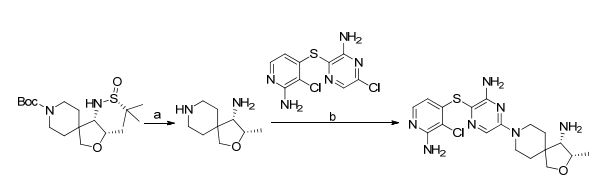
Step a: A mixture of (3S,4S)-tert-butyl 4-((R)-1,1-dimethylethylsulfinamido)-3-methyl-2-oxa-8- azaspiro[4.5]decane-8-carboxylate (51 mg, 0.136 mmol) and HCl (4 M in dioxane, 340 L, 1.362 mmol) in MeOH (5 mL) was stirred for 1 h at 40 °C. After cooling to RT, the volatiles were removed under reduced pressure to give (3S,4S)-3-methyl-2-oxa-8-azaspiro[4.5]decane-4-amine which was used in next step without further purification. MS m/z 171.1 (M+H)+. Step b: A mixture of (3S,4S)-3-methyl-2-oxa-8-azaspiro[4.5]decane-4-amine crude, 3-((2-amino3-chloropyridin-4-yl)thio)-6-chloropyrazin-2-amine (35.5 mg, 0.123 mmol), and DIPEA (193 L, 1.11 mmol) in DMSO (600 L) was stirred for 16 h at 100 °C. After cooling to RT, the volatiles were removed under reduced pressure and the resulting residue was purified by HPLC (gradient elution 15-40% acetonitrile in water, 5 mM NH4OH modifier) to give (3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (11 mg, 0.026 mmol). 1 H NMR (400 MHz, METHANOL-d4) δ ppm 7.67-7.47 (m, 2 H), 5.91 (d, J=5.5 Hz, 1 H), 4.22 (qd, J=6.4, 4.8 Hz, 1 H), 4.03 (ddt, J=13.5, 8.9, 4.7 Hz, 2 H), 3.86 (d, J=8.7 Hz, 1 H), 3.71 (d, J=8.7 Hz, 1 H), 3.37 (td, J=9.9, 4.9 Hz, 1 H), 3.29-3.23 (m, 1 H), 3.00 (d, J=5.0 Hz, 1H) 1.91-1.56 (m, 4 H), 1.21 (d, J=6.4 Hz, 3 H). HRMS calcd for C18H25ClN7OS (M+H)+ 422.1530, found 422.1514.
//////////TNO 155, CANCER