















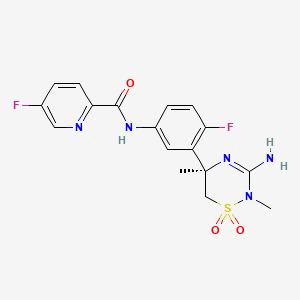
Verubecestat (MK-8931)
Merck Alzheimer’s drugs Verubecestat (MK-8931) is an oral β- amyloid precursor protein cleaving enzyme (BACE1 or β-secretase enzyme) inhibitor, is currently in Phase III clinical trials
Verubecestat
MK 8931, MK-8931, SCH 900931
2-Pyridinecarboxamide, N- (3 – ((5R) -3-amino-5,6-dihydro-2,5-dimethyl-1 , 1-dioxido-2H-1,2,4-thiadiazin-5-yl) -4-fluorophenyl) -5-fluoro-
N-[3-[(5R)-3-amino-2,5-dimethyl-1,1-dioxo-6H-1,2,4-thiadiazin-5-yl]-4-fluorophenyl]-5-fluoropyridine-2-carboxamide
CAS : 1286770-55-5
C17 H17 F2 N5 O3 S, 409.41
Mechanism: Oral β- amyloid precursor protein cleavage enzyme (BACE) inhibitors
Indications: Alzheimer’s disease
Development progress: phase III clinical
Companies: Merck
Verubecestat (MK-8931) is a small-molecule inhibitor of beta-secretase cleaving enzyme (BACE) 1 and BACE2 in development by Merck for the treatment of Alzheimer’s Disease.
MK-8931 is a beta-secretase 1 (BACE1) inhibitor in phase III development for the treatment of amnestic mild cognitive impairment (aMCI) due to Alzheimer’s disease at Merck & Co. The company is also conducting phase II/III trials for the treatment of Alzheimer’s type dementia.
Smiles: C [C @] 1 (CS (= O) (= O) N (C (= N1) N) C) c2cc (ccc2F) NC (= O) c3ccc (cn3) F
COSY PREDICT


https://www.google.co.in/patents/CN102639135A?cl=en
Scheme 3b:

The amine A (Scheme 3a, step 4) (13.7 g) in n-butanol (150 mL) was added a slurry solution of cyanogen bromide (5M, in MeCN). The resulting mixture was heated to reflux for 4 hours. The mixture was concentrated to 1/3 of original volume. To this mixture was added Et20 (200 mL). The resulting solid was removed by filtration, and the solid was washed with Et20 (2x). The solid was partitioned between EtOAc and saturated Na2CO3 (aq). The aqueous layer was extracted with EtOAc (3x). The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated to give 10.6 g
Scheme 10:

The nitro compound (Scheme 3b) (2. 50 g, 6. 0 mmol) of Et0H (150 mL) was degassed (To this solution was bubbled with nitrogen time 3 min). To this solution was added Pd / C (10% w / w, 50% water, 698 mg). The mixture was placed in a nitrogen atmosphere. Exhaust, and backfilled with H2 (3x). The obtained mixture at room temperature, followed by stirring under H2 balloon for 2 hours. Bubbling nitrogen gas, and the mixture was purged, filtered through Celite, and concentrated.Small plug filtered through a silica gel column, eluting with EtOAc, and the product was purified to give the aniline (2. 2g, 97%).
SEE
PATENT
http://www.google.co.in/patents/WO2011044181A1?cl=en

SNAPSHOT

SYNTHESIS CONSTUCTION
 AND
AND
 ON RXN WITH WITH BuLi GIVES
ON RXN WITH WITH BuLi GIVES
 THIS GIVES
THIS GIVES
 THIS ON TREATMENT WITH BrCN
THIS ON TREATMENT WITH BrCN
 ON BOC2O TREATMENT GIVES
ON BOC2O TREATMENT GIVES
 GIVES ON HYDGN
GIVES ON HYDGN

REACTION WITH

GIVES
FINAL COMPD Verubecestat

1H NMR PREDICT



13C NMR PREDICT


Updated…….WATCH OUT FOR MORE
https://www.google.co.in/patents/US8729071?cl=en

Steps 1-4:
These steps were performed using similar procedures to those described in steps 1-4 of Scheme 1a.
Step 5:
To a solution of the amine from step 4 (10.5 g, 36 mmol) in CH2Cl2 (200 mL) was added benzoylisothiocyanate (4.3 mL, 1.1 eq.). The resulting solution was stirred at RT for 2.5 days. Additional benzoylisothiocyanate (0.86 mL, 0.2 eq.) was added and the solution was stirred at RT for an additional 2 hours. The solution was then concentrated in vacuo.
A portion of this material (6.5 g, ˜14 mmol) was dissolved in MeOH (200 mL). To this solution was added Na2CO3 (s) (1.52 g, 14 mmol). The resultant mixture was stirred at RT for 45 min. After that time, a slight excess of HOAc was added to the solution. The mixture was then concentrated. The residue was partitioned between CH2Cl2 and ½ sat. NaHCO3 (aq.). The aqueous layer was extracted with CH2Cl2 (3×). The combined organic layers were dried over Na2SO4, filtered and concentrated. The thiourea (˜4.9 g) was carried onto the next reaction without further purification.
Step 6:
Example 15 was prepared using a method similar to that described in Scheme 1a step 6.

To a shiny of amine A (Scheme 3a step 4) (13.7 grams) in n-butanol (150 mL) was added a solution of cyanogen bromide (5M in MeCN). The resultant mixture was heated to reflux for 4 hours. The mixture was concentrated to ⅓ of the original volume. To the mixture was added Et2O (200 mL). The resultant solid was removed via filtration and the solid was washed with Et2O (2×). The solid was partitioned between EtOAc and sat. Na2CO3 (aq.). The aqueous layer was extracted with EtOAc (3×). The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated to afford 10.6 grams of Ex. 15. This material was converted to the t-butyl carbamate using a procedure similar to that described in Scheme 3.
Step 7:
A mixture of the bromide (3.00 g, 6.92 mmol), benzophenone imine (1.39 mL, 8.30 mmol), Pd2(dba)3 (0.634 g, 0.692 mmol), John-Phos (0.413 g, 1.38 mmol), sodium tert-butoxide (2.13 g, 22.1 mmol), and toluene (51 mL) was degassed (vacuum/N2). The mixture was then stirred at 65° C. under nitrogen for 3 h. After this time, the reaction mixture was cooled to room temperature and filtered through a pad of Celite and rinsed with ethyl acetate (100 mL). The filtrate was concentrated under reduced pressure. The residue was then dissolved in methanol (76 mL) and the resulting solution was charged with hydroxyl amine hydrochloride (2.16 g, 31.1 mmol) and sodium acetate (2.55 g, 31.1 mmol). The reaction mixture was stirred at room temperature for 40 min. After this time, the reaction mixture was concentrated under reduced pressure. The resulting residue was dissolved in ethyl acetate (200 mL) and washed with saturated aqueous sodium bicarbonate (100 mL), water (100 mL), and brine (100 mL). The organic layer was then dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography (silica, 0-100% ethyl acetate/heptane) to afford the amino pyridine (0.880 g, 34%).

To a flame-dried flask was added a pyridyl bromide (Table IIb, Entry 15, 1.5 g, 3.3 mmol), Pd2(dba)3 (305 mg, 0.3 mmol), (2-biphenyl)di-tert-butylphosphine (200 mg, 0.7 mmol), sodium tert-butoxide (1.02 g, 0.011 mmol), benzophenone imine (670 ul, 4 mmol), and toluene (21 mL). The mixture was evacuated under vacuum and back-filled with N2 (3×). The mixture was stirred at 60° C. for 1 h. After filtration through celite, the filtrate was concentrated. The crude residue was dissolved in 36 mL of methanol, and hydroxyl amine hydrochloride (458 mg, 6.6 mmol) and sodium acetate (541 mg, 6.6 mmol) were added. The reaction was stirred for 35 min and then quenched with saturated aqueous sodium bicarbonate. The mixture was extracted with ethyl acetate, and the combined organic portions were dried over magnesium sulfate and concentrated. The crude residue was purified by a flash silica column (50% ethyl acetate/hexane) to get an aminopyridine product (730 mg, 68%).

A solution of the nitro compound (Scheme 3b) (2.50 g, 6.0 mmol) in EtOH (150 mL) was degassed by bubbling N2 through the solution for 3 min. To this solution was added Pd/C (10% w/w, 50% H2O, 698 mg.). The mixture was placed under an atmosphere of N2. The atmosphere was evacuated and back-filled with H2 (3×). The resulting mixture was stirred at RT under a H2 balloon for 2 h. The mixture was purged by bubbling N2 through it, filtered through Celite and concentrated. The product was purified by filtering through a small plug of silica gel column eluting with EtOAc to afford the aniline (2.2 g, 97%).
| ENTRY 25 |
 MH+: 410.0, HPLC1.79 min, LCMSMETHOD D |
Method D:
- Column: Agilent Zorbax SB-C18 (3.0×50 mm) 1.8 uM
Mobile phase: A: 0.05% Trifluoroacetic acid in water
-
- B: 0.05% Trifluoroacetic acid in acetonitrile
Gradient: 90:10 (A:B) for 0.3 min, 90:10 to 5:95 (A:B) over 1.2 min, 5:95 (A:B) for 1.2 min.
Flow rate: 1.0 mL/min
UV detection: 254 and 220 nm
Mass spectrometer: Agilent 6140 quadrupole
update…………..
Discovery of the 3-Imino-1,2,4-thiadiazinane 1,1-Dioxide Derivative Verubecestat (MK-8931)–A β-Site Amyloid Precursor Protein Cleaving Enzyme 1 Inhibitor for the Treatment of Alzheimer’s Disease
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.
Abstract

Verubecestat 3 (MK-8931), a diaryl amide-substituted 3-imino-1,2,4-thiadiazinane 1,1-dioxide derivative, is a high-affinity β-site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitor currently undergoing Phase 3 clinical evaluation for the treatment of mild to moderate and prodromal Alzheimer’s disease. Although not selective over the closely related aspartyl protease BACE2, verubecestat has high selectivity for BACE1 over other key aspartyl proteases, notably cathepsin D, and profoundly lowers CSF and brain Aβ levels in rats and nonhuman primates and CSF Aβ levels in humans. In this annotation, we describe the discovery of 3, including design, validation, and selected SAR around the novel iminothiadiazinane dioxide core as well as aspects of its preclinical and Phase 1 clinical characterization.
N-[3-[(5R)-3-Amino-5,6-dihydro-2,5-dimethyl-1,1-dioxido-2H-1,2,4-thiadiazin-5-yl]-4-fluorophenyl]-5-fluoro-2-pyridinecarboxamide (3)
References
-
(a) Wortmann, M.Dementia: a global health priority – highlights from an ADI and World Health Organization report Alzheimer’s Res. Ther. 2012, 4, 40– 43, DOI: 10.1186/alzrt143
(b) Alzheimer’s Disease International. World Alzheimer Report 2015. http://www.alz.co.uk/research/WorldAlzheimerReport2015.pdf.
-
Alzheimer’s Association. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 2015, 332– 384.
-
Zemek, F.; Drtinova, L.; Nepovimova, E.; Sepsova, V.; Korabecny, J.; Klimes, J.; Kuca, K.Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine Expert Opin. Drug Saf.2014, 13, 759– 774, DOI: 10.1517/14740338.2014.914168
-
Haass, C.; Selkoe, D. J.Soluble protein oligomers in neurodegeneration: Lesson from the Alzheimer’s amyloid β-peptide Nat. Rev. Mol. Cell Biol. 2007, 8, 101– 112, DOI: 10.1038/nrm2101
-
(a) Hardy, J.; Selkoe, D. J.The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics Science 2002, 297, 353– 356, DOI: 10.1126/science.1072994
(b) Karran, E.; Mercken, M.; De Strooper, B.The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics Nat. Rev. Drug Discovery 2011, 10, 698– 712, DOI: 10.1038/nrd3505
Tanzi, R. E.The genetics of Alzheimer disease Cold Spring Harbor Perspect. Med. 2012, 2, 1– 10, DOI: 10.1101/cshperspect.a006296
-
Citron, M.Alzheimer’s disease: strategies for disease modification Nat. Rev. Drug Discovery 2010, 9, 387– 398, DOI: 10.1038/nrd2896
-
Jonsson, T.; Atwal, J. K.; Steinberg, S.; Snaedal, J.; Jonsson, P. V.; Bjornsson, S.; Stefansson, H.; Sulem,P.; Gudbjartsson, D.; Maloney, J.; Hoyte, K.; Gustafson, A.; Liu, Y.; Lu, Y.; Bhangale, T.; Graham, R. R.; Huttenlocher, J.; Bjornsdottir, G.; Andreassen, O. A.; Jönsson, E. G.; Palotie, A.; Behrens, T. W.; Magnusson, O. T.; Kong, A.; Thorsteinsdottir, U.; Watts, R. J.; Stefansson, K.A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline Nature 2012, 488, 96– 99, DOI: 10.1038/nature11283
-
Roberds, S. L.; Anderson, J.; Basi, G.; Bienkowski, M. J.; Branstetter, D. G.; Chen, K. S.; Freedman, S. B.; Frigon, N. L.; Games, D.; Hu, K.; Johnson-Wood, K.; Kappenman, K. E.; Kawabe, T. T.; Kola, I.; Kuehn,R.; Lee, M.; Liu, W.; Motter, R.; Nichols, N. F.; Power, M.; Robertson, D. W.; Schenk, D.; Schoor, M.; Shopp, G. M.; Shuck, M. E.; Sinha, S.; Svensson, K. A.; Tatsuno, G.; Tintrup, H.; Wijsman, J.; Wright, S.; McConlogue, L.BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeutics Hum. Mol. Genet. 2001, 10, 1317– 1324
-
Vassar, R.; Kuhn, P.-H.; Haass, C.; Kennedy, M. E.; Rajendran, L.; Wong, P. C.; Lichtenthaler, S. F.Function, therapeutic potential and cell biology of BACE proteases: current status and future prospectsJ. Neurochem. 2014, 130, 4– 28, DOI: 10.1111/jnc.12715
(a) Vassar, R.; Bennett, B. D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E. A.; Denis, P.; Teplow, D. B.; Ross, S.; Amarante, P.; Loeloff, R.; Luo, Y.; Fisher, S.; Fuller, J.; Edenson, S.; Lile, J.; Jarosinski, M. A.; Biere, A. L.; Curran, E.; Burgess, T.; Louis, J.-C.; Collins, F.; Treanor, J.; Rogers, G.; Citron, M.β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE Science 1999,286, 735– 741
(b) Sinha, S.; Anderson, J. P.; Barbour, R.; Basi, G. S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H. F.; Frigon, N.; Hong, J.; Jacobson-Croak, K.; Jewett, N.; Keim, P.; Knops, J.; Lieberburg, I.; Power, M.; Tan, H.; Tatsuno, G.; Tung, J.; Schenk, D.; Seubert, P.; Suomensaari, S. M.; Wang, S.; Walker, D.; Zhao, J.; McConlogue, L.; John, V.Purification and cloning of amyloid precursor protein β-secretase from human brain Nature 1999, 402, 537– 540, DOI: 10.1038/990114
(c) Hussain, I.; Powell, D.; Howlett, D. R.; Tew, D. G.; Meek,T. D.; Chapman, C.; Gloger, I. S.; Murphy, K. E.; Southan, C. D.; Ryan, D. M.; Smith, T. S.; Simmons, D. L.; Walsh, F. S.; Dingwall, C.; Christie, G.Identification of a novel aspartic protease (Asp 2) as β-SecretaseMol. Cell. Neurosci. 1999, 14, 419– 427, DOI: 10.1006/mcne.1999.0811
(d) Yan, R.; Bienkowski, M. J.; Shuck, M. E.; Miao, H.; Tory, M. C.; Pauley, A. M.; Brashler, J. R.; Stratman, N. C.; Mathews, W. R.; Buhl, A. E.; Carter, D. B.; Tomasselli, A. G.; Parodi, L. A.; Heinrikson, R. L.; Gurney,M. E.Membrane-anchored aspartyl protease with Alzheimer’s disease β-secretase activity Nature 1999,402, 533– 537, DOI: 10.1038/990107
(e) Lin, X.; Koelsch, G.; Wu, S.; Downs,D.; Dashti, A.; Tang, J.Human aspartic protease memapsin 2 cleaves the β-secretase site of β-amyloid precursor protein Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 1456– 1460, DOI: 10.1073/pnas.97.4.1456
-
(a) Oehlrich, D.; Prokopcova, H.; Gijsen, H. J. M.The evolution of amidine-based brain penetrant BACE1 inhibitors Bioorg. Med. Chem. Lett. 2014, 24, 2033– 2045, DOI: 10.1016/j.bmcl.2014.03.025
(b) Yuan, J.; Venkatraman, S.; Zheng, Y.; McKeever, B. M.; Dillard, L. W.; Singh, S. B.Structure-based design of β-site APP cleaving enzyme 1 (BACE) inhibitors for the treatment of Alzheimer’s disease. disease J. Med. Chem. 2013, 56, 4156– 4180, DOI: 10.1021/jm301659n
(c) Probst, G.; Xu, Y. z.Small-molecule BACE1 inhibitors: a patent literature review (2006 – 2011) Expert Opin. Ther. Pat. 2012, 22, 511– 540, DOI: 10.1517/13543776.2012.681302
-
(a) Willem, M.; Garratt, A. N.; Novak, B.; Citron, M.; Kaufmann, S.; Rittger, A.; DeStrooper, B.; Saftig, P.; Birchmeier, C.; Haass, C.Control of peripheral nerve myelination by the β-Secretase BACE1 Science2006, 314, 664– 666, DOI: 10.1126/science.1132341
(b) Hu, X.; Hicks, C. W.; He, W.; Wong, P.; Macklin, W. B.; Trapp, B. D.; Yan, R.Bace1 modulates myelination in the central and peripheral nervous system Nat. Neurosci. 2006, 9, 1520– 1525, DOI: 10.1038/nn1797
-
Cheret, C.; Willem, M.; Fricker, F. R.; Wende, H.; Wulf-Goldenberg, A.; Tahirovic, S.; Nave, K.-A.; Saftig,P.; Haass, C.; Garratt, A. N.; Bennett, D. L.; Birchmeier, C.Bace1 and Neuregulin-1 cooperate to control formation and maintenance of muscle spindles EMBO J. 2013, 32, 2015– 2028, DOI: 10.1038/emboj.2013.146
-
McConlogue, L.; Buttini, M.; Anderson, J. P.; Brigham, E. F.; Chen, K. S.; Freedman, S. B.; Games, D.; Johnson-Wood, K.; Lee, M.; Zeller, M.; Liu, W.; Motter, R.; Sinha, S.Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice J. Biol. Chem. 2007,282, 26326– 26334, DOI: 10.1074/jbc.M611687200
Wang, Y.-S.; Strickland, C.; Voigt, J. H.; Kennedy, M. E.; Beyer, B. M.; Senior, M. M.; Smith, E. M.; Nechuta, T. L.; Madison, V. S.; Czarniecki, M.; McKittrick, B. A.; Stamford, A. W.; Parker, E. M.; Hunter, J. C.; Greenlee, W. J.; Wyss, D. F.Application of fragment-based NMR screening, X-ray crystallography, structure-based design, and focused chemical library design to identify novel μM leads for the development of nM BACE-1 (β-site APP cleaving enzyme 1) inhibitors J. Med. Chem. 2010, 53, 942– 950, DOI: 10.1021/jm901472u
-
The CAS name represents the 3-aminothiadiazine tautomer, and the structures depicted in the manuscript represent the 3-iminothiadiazinane tautomer.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE



 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SON
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
///////