














, click to zoom.")
Vibegron, MK-4618, KRP 114V
Target-based Actions Beta 3 adrenoceptor agonist
Indications Overactive bladder; Urinary incontinence
UPDATE 2018/9/21 pmda Beova JAPAN 2018Kyorin Pharmaceutical, under license from Merck, is developing vibegron (phase II, September 2014) for the treating of overactive bladder. In July 2014, Merck has granted to Kyorin an exclusive license to develop, manufacture and commercialize vibegron in Japan.
MK-4618 is being developed in phase II clinical trials at Merck & Co. for the treatment of overactive bladder. The company had been developing the compound for the treatment of endocrine disorders and hypertension; however, recent progress reports are not available at present.
In 2014, Merck licensed the product to Kyorin for development and commercialization in Japan.
The function of the lower urinary tract is to store and periodically release urine. This requires the orchestration of storage and micturition reflexes which involve a variety of afferent and efferent neural pathways, leading to modulation of central and peripheral neuroeffector mechanisms, and resultant coordinated regulation of sympathetic and parasympathetic components of the autonomic nervous system as well as somatic motor pathways. These proximally regulate the contractile state of bladder (detrusor) and urethral smooth muscle, and urethral sphincter striated muscle.
β Adrenergic receptors (βAR) are present in detrusor smooth muscle of various species, including human, rat, guinea pig, rabbit, ferret, dog, cat, pig and non-human primate. However, pharmacological studies indicate there are marked species differences in the receptor subtypes mediating relaxation of the isolated detrusor; β1AR predominate in cats and guinea pig, β2AR predominate in rabbit, and β3AR contribute or predominate in dog, rat, ferret, pig, cynomolgus and human detrusor. Expression of βAR subtypes in the human and rat detrusor has been examined by a variety of techniques, and the presence of β3AR was confirmed using in situ hybridization and/or reverse transcription-polymerase chain reaction (RT-PCR). Real time quantitative PCR analyses of β1AR, β2AR and β3AR mRNAs in bladder tissue from patients undergoing radical cystectomy revealed a preponderance of β3AR mRNA (97%, cf 1.5% for β1AR mRNA and 1.4% for β2AR mRNA). Moreover, β3AR mRNA expression was equivalent in control and obstructed human bladders. These data suggest that bladder outlet obstruction does not result in downregulation of β3AR, or in alteration of β3AR-mediated detrusor relaxation. β3AR responsiveness also has been compared in bladder strips obtained during cystectomy or enterocystoplasty from patients judged to have normal bladder function, and from patients with detrusor hyporeflexia or hyperreflexia. No differences in the extent or potency of β3AR agonist mediated relaxation were observed, consistent with the concept that the β3AR activation is an effective way of relaxing the detrusor in normal and pathogenic states.
Functional evidence in support of an important role for the β3AR in urine storage emanates from studies in vivo. Following intravenous administration to rats, the rodent selective β3AR agonist CL316243 reduces bladder pressure and in cystomeric studies increases bladder capacity leading to prolongation of micturition interval without increasing residual urine volume.
Overactive bladder is characterized by the symptoms of urinary urgency, with or without urgency urinary incontinence, usually associated with frequency and nocturia. The prevalence of OAB in the United States and Europe has been estimated at 16 to 17% in both women and men over the age of 18 years. Overactive bladder is most often classified as idiopathic, but can also be secondary to neurological condition, bladder outlet obstruction, and other causes. From a pathophysiologic perspective, the overactive bladder symptom complex, especially when associated with urge incontinence, is suggestive of detrusor overactivity. Urgency with or without incontinence has been shown to negatively impact both social and medical well-being, and represents a significant burden in terms of annual direct and indirect healthcare expenditures. Importantly, current medical therapy for urgency (with or without incontinence) is suboptimal, as many patients either do not demonstrate an adequate response to current treatments, and/or are unable to tolerate current treatments (for example, dry mouth associated with anticholinergic therapy). Therefore, there is need for new, well-tolerated therapies that effectively treat urinary frequency, urgency and incontinence, either as monotherapy or in combination with available therapies. Agents that relax bladder smooth muscle, such as β3AR agonists, are expected to be effective for treating such urinary disorders.
PATENT
http://www.google.com/patents/WO2013062881A1?cl=en

EXAMPLE 3

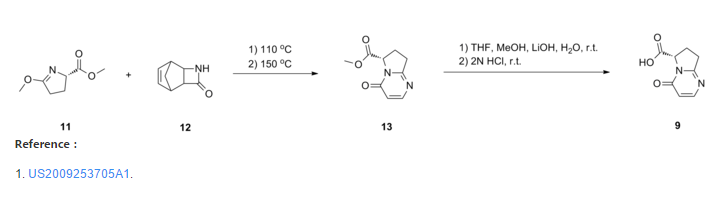
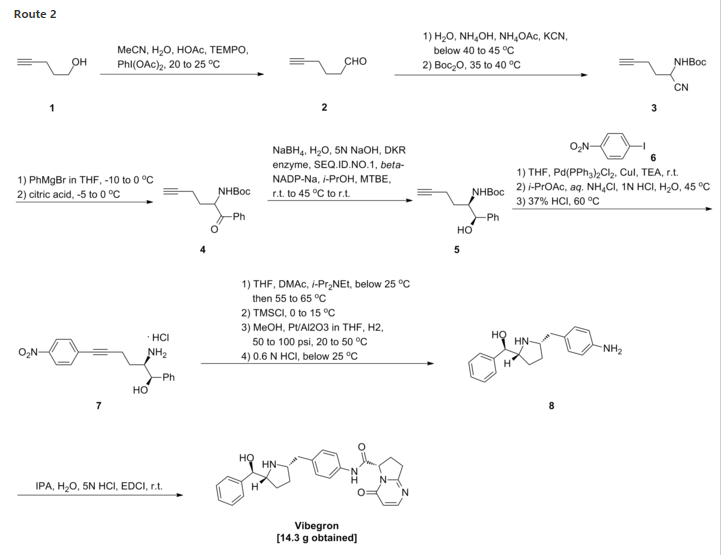
To a three neck flask equipped with a N2 inlet, a thermo couple probe was charged pyrrolidine i-11 (10.0 g), sodium salt i-12 (7.87 g), followed by IPA (40 mL) and water (24 mL). 5 N HC1 (14.9 mL) was then slowly added over a period of 20 min to adjust pH = 3.3- 3.5, maintaining the batch temperature below 35 °C. Solid EDC hydrochloride (7.47 g) was charged in portions over 30 min. The reaction mixture was aged at RT for additional 0.5 – 1 h, aqueous ammonia (14%) was added dropwise to pH ~8.6. The batch was seeded and aged for additional 1 h to form a slurry bed. The rest aqueous ammonia (14%, 53.2 ml total) was added dropwise over 6 h. The resulting thick slurry was aged 2-3 h before filtration. The wet-cake was displacement washed with 30% IPA (30 mL), followed by 15% IPA (2 x 20mL) and water (2 X 20mL). The cake was suction dried under N2 overnight to afford 14.3 g of compound of Formula (I)-
1H NMR (DMSO) δ 10.40 (s, NH), 7.92 (d, J = 6.8, 1H), 7.50 (m, 2H), 7.32 (m, 2H), 7.29 (m, 2H), 7.21 (m, 1H), 7.16 (m, 2H), 6.24 (d, J = 6.8, 1H), 5.13 (dd, J = 9.6, 3.1, 1H), 5.08 (br s, OH), 4.22 (d, J = 7.2, 1H), 3.19 (p, J = 7.0, 1H), 3.16-3.01 (m, 3H), 2.65 (m, 1H), 2.59-2.49 (m, 2H), 2.45 (br s, NH), 2.16 (ddt, J = 13.0, 9.6, 3.1, 1H), 1.58 (m, 1H), 1.39 (m, 1H), 1.31-1.24 (m, 2H).
13C NMR (DMSO) δ 167.52, 165.85, 159.83, 154.56, 144.19, 136.48, 135.66, 129.16, 127.71, 126.78, 126.62, 119.07, 112.00, 76.71, 64.34, 61.05, 59.60, 42.22, 31.26, 30.12, 27.09, 23.82.
HPLC method – For monitoring conversion
Column: XBridge C18 cm 15 cm x 4.6 mm, 3.5 μιη particle size;
Column Temp. : 35 °C; Flow rate: 1.5 mL/min; Detection: 220 nm;
Mobile phase: A. 5 mM Na2B407.10 H20 B: Acetonitrile
Gradient:

HPLC method – For level of amide epimer detection
Column: Chiralpak AD-H 5 μηι, 250 mm x 4.6 mm.
Column Temp: 35 °C; Flow rate: 1.0 mL/min; Detection: 250 nm;
Mobile phase: Isocratic 30% Ethanol in hexanes + 0.1% isobutylamine
PATENT
WO 2009124167
http://www.google.com/patents/WO2009124167A1?cl=en
EXAMPLE 103
(6y)-N-r4-({(‘25′. 5R)-5-r(‘R)-hvdroxy(‘phenvnmethyl1pyrrolidin-2-yl}methvnphenyl1-4-oxo- 4,6J,8-tetrahydropyiτolori,2-α1pyrimidine-6-carboxamide

ter?-butyl(2R. 55f)-2-rCR)-hvdroxy(‘phenvnmethyl1-5-r4-(‘{r(‘65f)-4-oxo-4.6.7.8-
tetrahydropyrrolof 1.2-alpyrimidin-6- yl]carbonyl} amino)benzyl]pyrrolidine- 1 – carboxylate

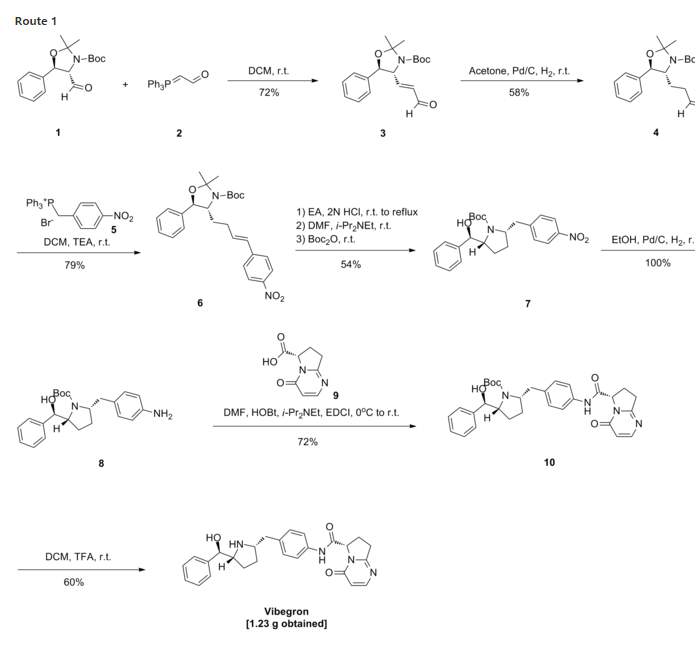
To a solution of i-13a (21.4 g, 55.9 mmol) in N,N-dimethylformamide (100 ml) at O0C was added [(65)-4-oxo-4,6,7,8-tetrahydropyrrolo[l,2-α]pyrimidine-6-carboxylic acid (11.1 g, 61.5 mmol), followed by 1 -hydroxybenzotriazole (i-44, 7.55 g, 55.9 mmol), N-(3- dimethylaminopropyl)-Nl-ethylcarbodiimide hydrochloride (16.1 g, 84.0 mmol) and N,N- diisopropylethylamine (29.2 ml, 168 mmol). The reaction mixture was stirred from O0C to ambient temperature for 2 h. Water (600 ml) was added and it was extracted with dichloromethane (600 ml x 2). The combined organic layers were dried over Na2SO4. After removal of the volatiles, the residue was purified by using a Biotage Horizon® system (0-5% then 5% methanol with 10% ammonia/dichloromethane mixture) to afford the title compound which contained 8% of the minor diastereomer. It was further purified by supercritical fluid chromatography (chiral AS column, 40% methanol) to afford the title compound as a pale yellow solid (22.0 g, 72%). 1H NMR (CDCl3): δ 9.61 (s, IH), 7.93 (d, J = 6.6 Hz, IH), 7.49 (d, J = 8.4 Hz, 2H), 7.35-7.28 (m, 5H), 7.13 (d, J = 8.5 Hz, 2H), 6.40 (d, J = 6.7 Hz, IH), 5.36 (d, J = 8.6 Hz, IH), 4.38 (m, IH), 4.12-4.04 (m, 2H), 3.46 (m,lH), 3.15-3.06 (m, 2H), 2.91 (dd, J = 13.1, 9.0 Hz, IH), 2.55 (m, IH), 2.38 (m, IH), 1.71-1.49 (m, 13H). LC-MS 567.4 (M+23).
(6S)-N-\4-( U2S. 5R)-5-r(R)-hvdroxy(phenyl)methyl1pyrrolidin-2-
yl}methyl)phenyl1-4-oxo-4,6J,8-tetrahvdropyrrolori,2-α1pyrimidine-6- carboxamide

To a solution of the intermediate from Step A (2.50 g, 4.59 mmol) in dichloromethane (40 ml) was added trifluoroacetic acid (15 ml). The reaction mixture was stirred at ambient temperature for 1.5 h. After removal of the volatiles, saturated NaHCCh was added to make the PH value to 8-9. The mixture was then extracted with dichloromethane. The combined organic layers were dried over Na2SO4. After concentration, crystallization from methanol/acetonitrile afforded the title compound as a white solid (1.23g, 60%). 1H NMR (DMSO-Cl6): δ 10.40 (s, IH), 7.91 (d, J = 6.7 Hz, IH), 7.49 (d, J = 8.3 Hz, 2H), 7.32-7.26 (m, 4H), 7.21 (m, IH), 7.15 (d, J = 8.4 Hz, 2H), 6.23 (d, J = 6.7 Hz, IH), 5.11 (dd, J = 9.6, 2.9 Hz, IH), 5.10 (br, IH), 4.21 (d, J = 7.1 Hz, IH), 3.20-3.00 (m, 4H), 2.66-2.51 (m, 3H), 2.16 (m, IH), 1.57 (m, IH), 1.38 (m, IH), 1.29-1.23 (m, 2H). LC-MS 445.3 (M+l).
Using the Biological Assays described above, the human β3 functional activity of Example 103 was determined to be between 11 to 100 nM.
PATENT
CHECK STRUCTURE…………….CAUTION
http://www.google.com/patents/US8247415


CAUTION…………….
Example 103(6S)-N-[4-({(2S,5R)-5-[(R)-hydroxy(phenyl)methyl]pyrrolidin-2-yl}methyl)phenyl]-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidine-6-carboxamide

Step A: tert-butyl(2R,5S)-2-[(R)-hydroxy(phenyl)methyl]-5-[4-({[(6S)-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidin-6-yl]carbonyl}amino)benzyl]pyrrolidine-1-carboxylate

To a solution of i-13a (21.4 g, 55.9 mmol) in N,N-dimethylformamide (100 ml) at 0° C. was added [(6S)-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidine-6-carboxylic acid (11.1 g, 61.5 mmol), followed by 1-hydroxybenzotriazole (i-44, 7.55 g, 55.9 mmol), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (16.1 g, 84.0 mmol) and N,N-diisopropylethylamine (29.2 ml, 168 mmol). The reaction mixture was stirred from 0° C. to ambient temperature for 2 h. Water (600 ml) was added and it was extracted with dichloromethane (600 ml×2). The combined organic layers were dried over Na2SO4. After removal of the volatiles, the residue was purified by using a Biotage Horizon® system (0-5% then 5% methanol with 10% ammonia/dichloromethane mixture) to afford the title compound which contained 8% of the minor diastereomer. It was further purified by supercritical fluid chromatography (chiral AS column, 40% methanol) to afford the title compound as a pale yellow solid (22.0 g, 72%). 1H NMR (CDCl3): δ 9.61 (s, 1H), 7.93 (d, J=6.6 Hz, 1H), 7.49 (d, J=8.4 Hz, 2H), 7.35-7.28 (m, 5H), 7.13 (d, J=8.5 Hz, 2H), 6.40 (d, J=6.7 Hz, 1H), 5.36 (d, J=8.6 Hz, 1H), 4.38 (m, 1H), 4.12-4.04 (m, 2H), 3.46 (m, 1H), 3.15-3.06 (m, 2H), 2.91 (dd, J=13.1, 9.0 Hz, 1H), 2.55 (m, 1H), 2.38 (m, 1H), 1.71-1.49 (m, 13H). LC-MS 567.4 (M+23).
Step B: (6S)-N-[4-({(2S,5R)-5-[(R)-hydroxy(phenyl)methyl]pyrrolidin-2-yl}methyl)phenyl]-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidine-6-carboxamide

To a solution of the intermediate from Step A (2.50 g, 4.59 mmol) in dichloromethane (40 ml) was added trifluoroacetic acid (15 ml). The reaction mixture was stirred at ambient temperature for 1.5 h. After removal of the volatiles, saturated NaHCO3 was added to make the PH value to 8-9. The mixture was then extracted with dichloromethane. The combined organic layers were dried over Na2SO4. After concentration, crystallization from methanol/acetonitrile afforded the title compound as a white solid (1.23 g, 60%). 1H NMR (DMSO-d6): δ 10.40 (s, 1H), 7.91 (d, J=6.7 Hz, 1H), 7.49 (d, J=8.3 Hz, 2H), 7.32-7.26 (m, 4H), 7.21 (m, 1H), 7.15 (d, J=8.4 Hz, 2H), 6.23 (d, J=6.7 Hz, 1H), 5.11 (dd, J=9.6, 2.9 Hz, 1H), 5.10 (br, 1H), 4.21 (d, J=7.1 Hz, 1H), 3.20-3.00 (m, 4H), 2.66-2.51 (m, 3H), 2.16 (m, 1H), 1.57 (m, 1H), 1.38 (m, 1H), 1.29-1.23 (m, 2H). LC-MS 445.3 (M+1).
Using the Biological Assays described above, the human β3 functional activity of Example 103 was determined to be between 11 to 100 nM.
PATENT
WO2014150639
Step 6. Preparation of Compound 1-7 from Compound 1-6 and Compound A-2

To a three neck flask equipped with a N2 inlet, a thermo couple probe was charged pyrrolidine hemihydrate 1-6 (10.3 g), sodium salt A-2 (7.87 g), followed by IPA (40 mL) and water (24 mL). 5 N HC1 (14.9 mL) was then slowly added over a period of 20 minutes to adjust pH = 3.3-3.5, maintaining the batch temperature below 35°C. Solid EDC hydrochloride (7.47 g) was charged in portions over 30 minutes. The reaction mixture was aged at RT for additional 0.5 – 1 hour, aqueous ammonia (14%) was added dropwise to pH -8.6. The batch was seeded and aged for additional 1 hour to form a slurry bed. The rest aqueous ammonia (14%, 53.2 ml total) was added dropwise over 6 hours. The resulting thick slurry was aged 2-3 hours before filtration. The wet-cake was displacement washed with 30% IPA (30 mL), followed by 15% IPA (2 x 20mL) and water (2 X 20mL). The cake was suction dried under N2 overnight to afford 14.3 g of compound 1-7.
1H NMR (DMSO) δ 10.40 (s, NH), 7.92 (d, J = 6.8, 1H), 7.50 (m, 2H), 7.32 (m, 2H), 7.29 (m, 2H), 7.21 (m, 1H), 7.16 (m, 2H), 6.24 (d, J = 6.8, 1H), 5.13 (dd, J = 9.6, 3.1, 1H), 5.08 (br s, OH), 4.22 (d, J = 7.2, 1H), 3.19 (p, J = 7.0, 1H), 3.16-3.01 (m, 3H), 2.65 (m, 1H), 2.59-2.49 (m, 2H), 2.45 (br s, NH), 2.16 (ddt, J = 13.0, 9.6, 3.1, 1H), 1.58 (m, 1H), 1.39 (m, 1H), 1.31-1.24 (m, 2H).
13C NMR (DMSO) δ 167.52, 165.85, 159.83, 154.56, 144.19, 136.48, 135.66, 129.16, 127.71, 126.78, 126.62, 119.07, 112.00, 76.71, 64.34, 61.05, 59.60, 42.22, 31.26, 30.12, 27.09, 23.82.
The crystalline freebase anhydrous form I of Compound 1-7 can be characterized by XRPD by

PATENT
WO-2014150633
Merck Sharp & Dohme Corp
Process for preparing stable immobilized ketoreductase comprises bonding of recombinant ketoreductase to the resin in a solvent. Useful for synthesis of vibegron intermediates. For a concurrent filling see WO2014150639, claiming the method for immobilization of ketoreductase. Picks up from WO2013062881, claiming the non enzymatic synthesis of vibegron and intermediates.
PAPER
Discovery of Vibegron: A Potent and Selective β3 Adrenergic Receptor Agonist for the Treatment of Overactive Bladder
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01372
http://pubs.acs.org/doi/suppl/10.1021/acs.jmedchem.5b01372/suppl_file/jm5b01372_si_001.pdf

The discovery of vibegron, a potent and selective human β3-AR agonist for the treatment of overactive bladder (OAB), is described. An early-generation clinical β3-AR agonist MK-0634 (3) exhibited efficacy in humans for the treatment of OAB, but development was discontinued due to unacceptable structure-based toxicity in preclinical species. Optimization of a series of second-generation pyrrolidine-derived β3-AR agonists included reducing the risk for phospholipidosis, the risk of formation of disproportionate human metabolites, and the risk of formation of high levels of circulating metabolites in preclinical species. These efforts resulted in the discovery of vibegron, which possesses improved druglike properties and an overall superior preclinical profile compared to MK-0634. Structure–activity relationships leading to the discovery of vibegron and a summary of its preclinical profile are described.
| Reference | ||
|---|---|---|
| 1 | H.P. Kaiser, et al., “Catalytic Hydrogenation of Pyrroles at Atmospheric Pressure“, J. Org. Chem., vol. 49, No. 22, p. 4203-4209 (1984). | |
ClinicalTrials.gov Web Site 2011, April 28
| WO2011043942A1 * | Sep 27, 2010 | Apr 14, 2011 | Merck Sharp & Dohme Corp. | Combination therapy using a beta 3 adrenergic receptor agonist and an antimuscarinic agent |
| US20090253705 * | Apr 2, 2009 | Oct 8, 2009 | Richard Berger | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| US20110028481 * | Apr 2, 2009 | Feb 3, 2011 | Richard Berger | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8642661 | Aug 2, 2011 | Feb 4, 2014 | Altherx, Inc. | Pharmaceutical combinations of beta-3 adrenergic receptor agonists and muscarinic receptor antagonists |
| US8653260 | Jun 20, 2012 | Feb 18, 2014 | Merck Sharp & Dohme Corp. | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| US20120202819 * | Sep 27, 2010 | Aug 9, 2012 | Merck Sharp & Dohme Corporation | Combination therapy using a beta 3 adrenergic receptor agonists and an antimuscarinic agent |
| US20020028835 | Jul 12, 2001 | Mar 7, 2002 | Baihua Hu | Cyclic amine phenyl beta-3 adrenergic receptor agonists |
| US20070185136 | Feb 2, 2007 | Aug 9, 2007 | Sanofi-Aventis | Sulphonamide derivatives, their preparation and their therapeutic application |
| US20110028481 | Apr 2, 2009 | Feb 3, 2011 | Richard Berger | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| WO2003072572A1 | Feb 17, 2003 | Sep 4, 2003 | Jennifer Anne Lafontaine | Beta3-adrenergic receptor agonists |
|
8-22-2012
|
Hydroxymethyl pyrrolidines as [beta]3 adrenergic receptor agonists
|
////////////C1CC(NC1CC2=CC=C(C=C2)NC(=O)C3CCC4=NC=CC(=O)N34)C(C5=CC=CC=C5)O